Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
Tudománytörténeti bevezetés A Mendel-i öröklődés 2015.

2
Gregor Johann Mendel (1822–1884)
Brünn – borsó palánta 1865. febr. 8. – előadás a Brünn-i Természettudományi Társaságban 1866. Mendel, G., Versuche über Pflanzen-Hybriden. (Verh. Naturforsch. Ver. Brünn 4: 3–47) 1900. Mendel munkássága jelentőségének felismerése (Hugo de Vries, Carl Correns és Erich von Tschermak)
 Mendel munkássága jelentőségének felismerése (Hugo de Vries, Carl Correns és Erich von Tschermak)")
3
Mendel a felismerések leírásakor
még NEM tudott: Kromoszómákról DNS-ről Génekről

4
MENDEL HIPOTÉZISE Mendel feltételezte a kromoszómák létezését
37 évvel azelőtt, hogy Sutton leírta volna őket. A jellegek hatásának leírásához Mendel statisztikai elemzést végzett Sutton mikroszkóppal írta le, hogy a kromoszómák hasonlóan viselkednek mint a medeli faktorok Párokban Gaméták képzésénél válnak el Meiózis során válnak el A párok képződése független MENDEL HIPOTÉZISE Mendel faktorai Kromoszómák

5
Tulajdonság Fenotípus Fehérje DNS / Gén Genotípus

6
Allélok Lókusz

7
Identikus allélok - egyazon gén két
allélja pontosan azonos Nem identikus allélok – amennyiben a fenotpus csupán hasonló Izoallélok - csak különleges környezeti tényezők között lehet megkülönböztetni Multiplex allélia - számos gén többszörös változatban létezik

8
Homozigóta Heterozigóta Hemizigóta X Y

9
Mendeli öröklődés szabályai
A fentotípus nem mindig követi a genotípusban adott jelleget Domináns jellegek képesek a recesszív jellegek hatásának elnyomására – ld. F1 nemzedék – heterozigóták A recesszív jelleg fenotípusos megjelenésének gyakorisága heterozigóták keresztezésekor 25% (ld. 3:1 arány) Homozigóta recesszívek keresztezésével a recesszív jelleg gyakorisága 100% marad
 Homozigóta recesszívek keresztezésével a recesszív jelleg. gyakorisága 100% marad.")
10
Több tulajdonság öröklődésének vizsgálata

11
Mendel harmadik szabálya - Független öröklődés - a kapcsolódási csoportokra csak korlátozottan érvényes

12
Inkomplet dominancia (Intermedier öröklődés)
Heterozigóták köztes fenotípust mutatnak

14
Autoszomális monogénes öröklődés
Dr. habil. Kőhidai László SE, Genetikai, Sejt- és Immunbiológiai Intézet Budapest 2015.

15
Autoszómális - Domináns
A tüneteket mutató személy legalább egyik szülője is érintett Homozigóta formában általában súlyosabb fenotípust eredményez Mindkét nembeli érintett lehet Ffi és nő egyaránt átörökít Beteg x Egészséges fenotípus > 50% lehet beteg Vertikális családfa Többnyire az apai életkor befolyásolja a mutáció gyakoriságát AD mutációk: receptor, szerkezeti vagy carrier fehérje mut. Variábilis expresszió és penetrancia

16
Domináns autoszómális
Kb ismert dominánsan öröklődő jelleg Átlagos gyakoriság 0.1-3/1000 szülés Gyakran érintett szerv/szervrendszerek: Csontváz Igedrendszer

17
Achondroplasia Gyakoriság 1:25000 FGFR3 gén mutációja
(fibroblast-growth factor receptor 3) A csöves csontok hossznövekedése károsodik Kéz és láb érintettség Magas homlok, középső arc gyengén fejl.
 A csöves csontok. hossznövekedése károsodik. Kéz és láb érintettség. Magas homlok, középső arc gyengén fejl.")
18
Achondroplasia

19
Rhinoceros unicornis házi juh anchon juh Teleoceras fossiger (Myocén-kori törpe rhino)
")
20
locus: 4p16.3 FGFR3 gén DNS: 16.5 Kb; 19 exon; exon 1 nem ismert emberben RNS: 4.0 Kb mRNS; alternatív splicing 7 és 8 exon esetében: két mRNS izoforma van IIIb és IIIc Expresszió: agy, porc, máj, vese, belső fül

21
Fehérje 806 as; 115 kDa funkció: tirozin kináz receptor szerkezet:
extracelluláris rész 3 Ig-szerű hurok (I, II, III) erősen hidrofób TM domén (22 as) -TM intracelluláris domén tirozin kináz aktivitással -TK
 erősen hidrofób TM domén (22 as) -TM. intracelluláris domén tirozin kináz aktivitással -TK.")
22
Az FGFR3 gén mutációi 3 öröklődő betegség is köthető FGFR3 mutációkhoz

23
Antoine Bernard-Jean Marfan
Arachnodactylia – Marfan szindróma Antoine Bernard-Jean Marfan (1896) Gabrielle
 Gabrielle.")
24
Arachnodactylia – Marfan szindróma
Tutankhamen fáraó Ehnaton fáraó

25
Skóciai Mária királynő
Abraham Lincoln

26
Marfan szindróma – Tünetek
csontok és izületek érintettsége testmagasság mellaks hosszú ujjak hiperflexibilitás

27
Az életkor előrehaladtával a mutációk gyakorisága nő
Marfan szindróma - Tünetek Szem és látás myopia, szemtengely hosszú lencse abnormális helyzetű szívbillentyű prolapszus (előesés) aorta aneurizma alacsony vérnyomás Szív és keringés Az életkor előrehaladtával a mutációk gyakorisága nő
 aorta aneurizma. alacsony vérnyomás. Szív és keringés. Az életkor előrehaladtával a mutációk gyakorisága nő.")
28
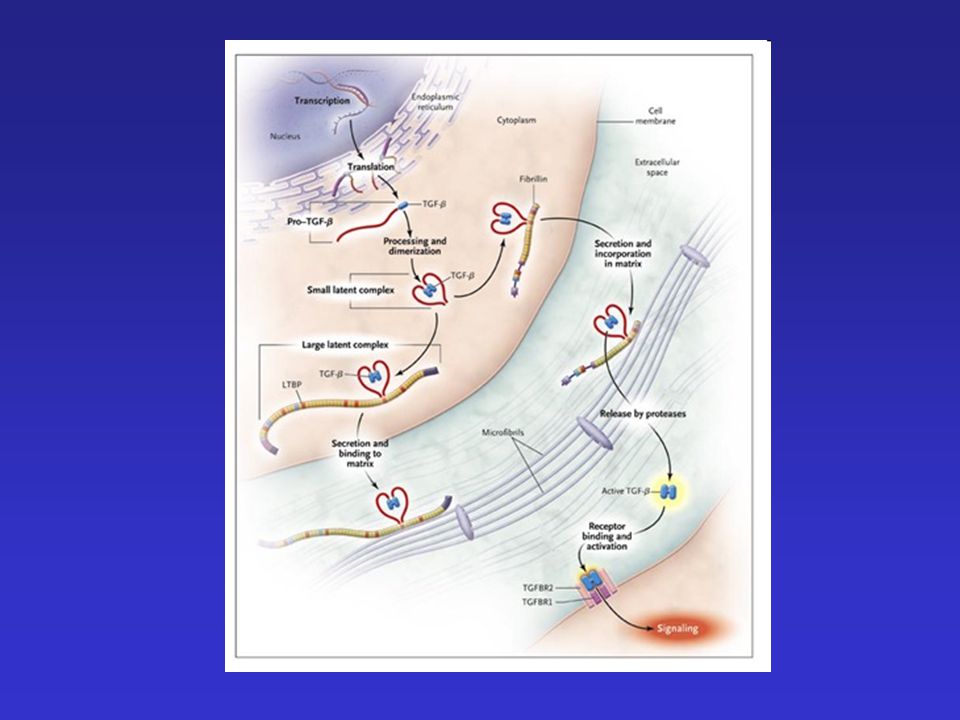
Marfan szindróma Fibrillin gén (FBN1) Fibrillin fehérje
15q21.1 A fibrillin gén számos mutációját írta le a szakirodalom (ld. zöld szín) Fibrillin fehérje kb. 60 domain 47 Ca2+-ot köt hasonlóság az EGF-hez
 Fibrillin fehérje. kb. 60 domain. 47 Ca2+-ot köt. hasonlóság az EGF-hez.")
31
Osteogenesis imperfecta I.
Kék sclera 100% penetrancia csonttörékenység süketség ill. hallásvesztés (általában nem 100%-os penetrancia) Magas fokú pleiotrópia
 Magas fokú pleiotrópia.")
32
Osteogenesis imperfecta
17q21.31-q22 COL1A1 gén COL1A kb 52 exon ( 6 – 49: alfa helikális domént kódolja) rövid exonok: 45 bp, 54 bp vagy ezek ismétlése RNS: 2 RNS: 5,8 kb és 4,8 kb eltérés: 3’ UTR Protein : 140 kDa
 rövid exonok: 45 bp, 54 bp vagy ezek ismétlése. RNS: 2 RNS: 5,8 kb és 4,8 kb eltérés: 3’ UTR. Protein : 140 kDa.")
33
Kollagén rost szerkezete
egészséges osteogenesis imp. I. típ. Centrális helikális domén: - 338 x ismétlődése a Gly-X-Y tripletnek - az X és Y gyakran prolin

34
Osteogenesis Imperfecta: kollagén mutációs térkép

35
Osteogenesis imperfecta

37
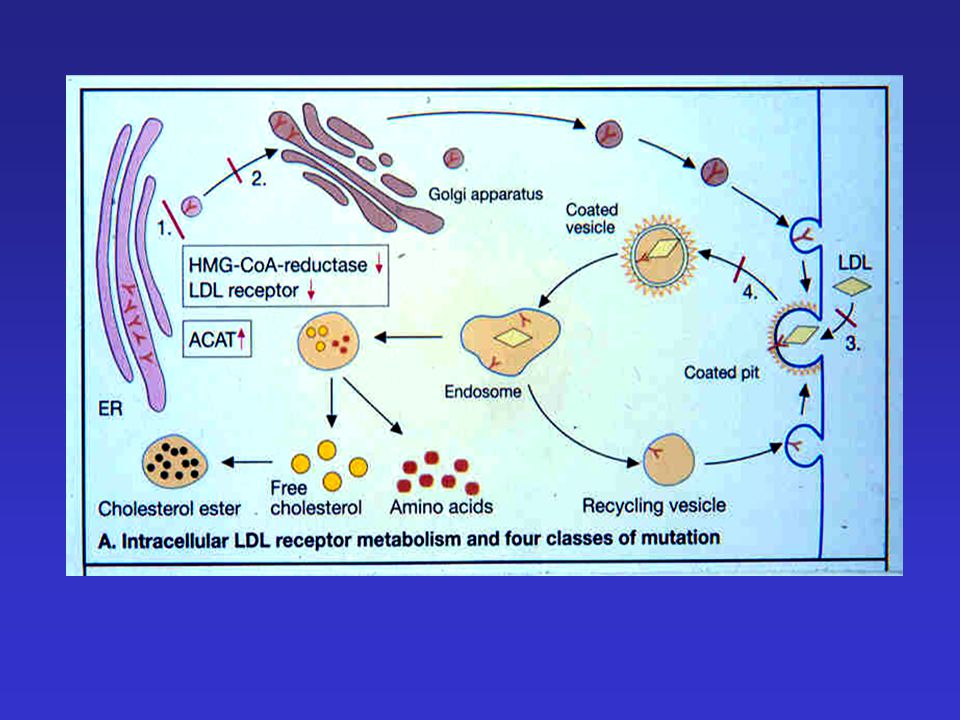
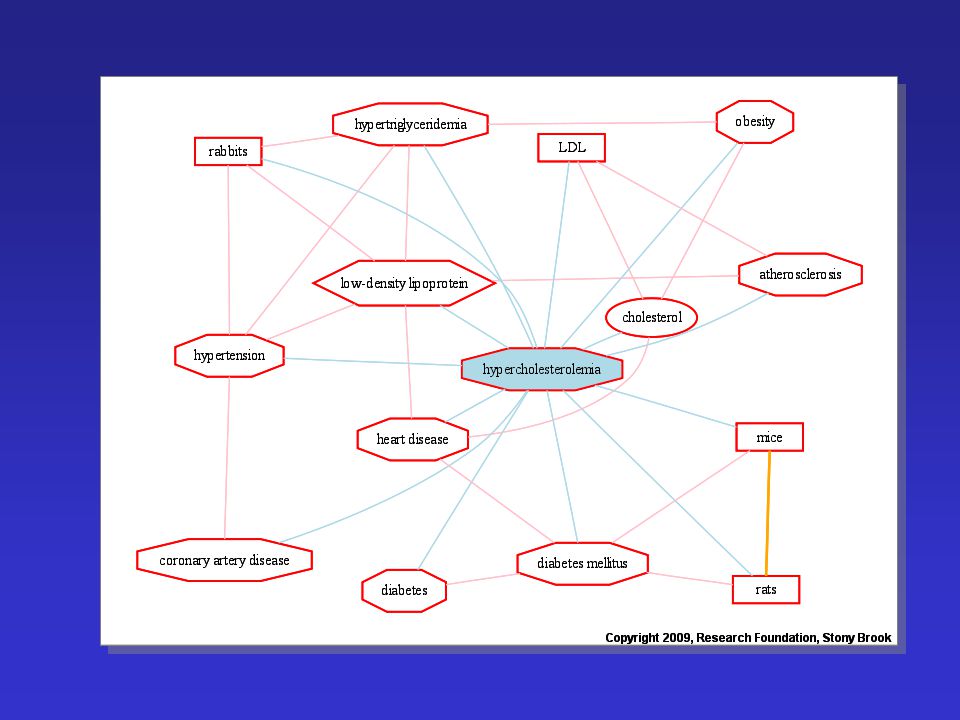
Familiáris hypercholesterinaemia
Vezető klinkai tünetek: - szív-érrendszeri betegségek korai előfordulása (szívinfarktus, agyi és perifériás érbetegségek) xanthoma szemtünetek
 xanthoma. szemtünetek.")
38
hypercholesterinaemia
Familiáris hypercholesterinaemia LDL életidő Egészséges: 2.5 nap FH: 4.5 nap Fokozott szérum LDL-szint Okai: - LDL-receptor mutáció - ApoB defektus LDL

40
19p

41
Familiáris hypercholesterinaemia
LDL-receptor mutáció Heterozygóta: 1: Homozygóta: 1: Leggyakoribb mutáció: 9. exon 408 kodon CTG → CTA Val → Met

42
Familiáris hypercholesterinaemia
LDL-receptor mutáció következményei Ligand kötő domain EGFP domain O-linked szénh.dom. Citopl. domain Membrán Joseph Goldstein, Michael Brown (Nobel díj 1985)
")
44
Trinukleotid-repeat betegségek

45
Huntington chorea OK: CAG trinukleotid repeat
A betegség éves kor között kezdődik Motoros, kognitív és pszichiátriai tünetek együttese CAG repeat szám: Normál - >26 Átmeneti 27-35 Alacsony penetrancia 36-39 Teljes penetrancia 40 felett OK: CAG trinukleotid repeat

46
Huntington chorea

47
Huntington chorea - (CAGn)
4 Funkció-nyeréses mutáció Huntingtin gén funkciója egészségesben nem ismert

48
Huntington chorea - Huntingtin

49
Huntington chorea – Érintett agyi központok

50
Huntington chorea Huntingtin hatása gén szinten
Dopamin D2 receptor gén expressziójának gátlása

51
Huntington chorea Huntingtin hatása sejtváz szinten
BDNF - brain-derived neurotrophic factor Review: Huntington disease is an Autosomally Dominant degenerative disorder resulting from expansion (>37 units) of a polyglutamine repeats in Huntingtin, a 350-kDa protein of unknown function. The polyglutamine repeat is localized in the N-terminal region of Huntingtin and is encoded by exon1 of the HD or Htt (Huntingtin) gene. Huntington disease is characterized by uncontrolled movements, personality changes, and dementia and causes the death of patients within 10–20 years after the appearance of the first symptoms. Huntingtin is highly expressed in the brain, and particularly enriched in Cerebral Cortex and Striatum. It is a cytoplasmic protein that is essential during development for Gastrulation and Neurogenesis, and it is important for Neuronal survival in the adult. Wild-type Huntingtin is anti-apoptotic in neurons in the Central Nervous System. Wild-type Huntingtin also reduces the toxicity of mutant Huntingtin in vivo. Huntingtin is also involved in vesicle trafficking in the secretory and endocytic pathways (Ref.1). Huntingtin shuttles between the nucleus and the cytoplasm. Full-length Huntingtin is predominantly distributed in the cytoplasm, whereas N-terminal fragments of Huntingtin with expanded polyglutamine tracts accumulate in the nucleus. Alternate Huntingtin conformations, are associated with cytoplasmic speckles and nuclear speckles or with perinuclear membranes and nucleoli. Moreover, its front end can associate with dozens of signal transduction proteins. This cargo includes members of transcription complexes [basal transcription factors (for example, SP1, TAFII130 (TBP-Associated Factor, RNA Polymerase II)), transcriptional co-repressors and co-activators (for example, NCOR1 (Nuclear Receptor Co-Repressor-1), Sin3A, CTBP (C-Terminal-Binding Protein), REST (RE1-Silencing Transcription Factor), p53), spliceosome and polyadenylation factors (for example, FBP11, symplekin)], a ubiquitin protein ligase (E2-25 kD) that regulates transcription factor turnover, effectors [receptors (for example, NMDAR (N-methyl-D-aspartate calcium channel receptors), EGFR (Epidermal Growth Factor Receptor)), Kinase (MLK2 (Mixed Lineage Kinase-2)), and Phosphatase (SHP2)], and adaptor proteins that localize signaling complexes (for example, GRB2 (Growth Factor Receptor-Bound Protein-2), PACSIN1 (Protein Kinase-C and Casein Kinase Substrate in Neurons-1), PSD95 (Postsynaptic Density-95), HAP1 (Huntingtin’s associated protein-1). Huntington disease pathogenesis can be viewed as a cascade of events that is first triggered by mutant Huntingtin through an abnormal interaction with an as-yet unknown cellular constituent. Once triggered, this cascade broadens with time, leading eventually to neuronal cell death (Ref.2). The Huntingtin’s expanded polyglutamine alters protein conformation, resulting in aberrant protein interactions, including interactions of the expanded polyglutamine with cellular proteins containing short polyglutamine stretches. CBP (CREB Binding Protein) is a co activator for CREB (cAMP Response Element-Binding Protein)-mediated transcription and contains an 18-glutamine stretch. CREB-mediated gene transcription promotes cell survival, and CBP is a major mediator of survival signals in mature neurons. CBP is a large protein with multiple domains and in addition to interacting with several different transcription factors also has a Histone Acetylase enzyme activity. The expanded polyglutamine in HD can interact with the short glutamine repeat in CBP, interfering with CBP function, causing transcriptional abnormalities, and leading to cellular toxicity. Mutant Huntingtin binds to CBP, drawing it into insoluble protein aggregates. Mutant Huntingtin also interacts directly with the acetyltransferase domain of CBP, blocking this activity. Activation of CBP acetyltransferase activity by transcriptional regulators results in the acetylation of histones in the promoter and enhancer regions of active genes, contributing to transcriptional activation by making these genes more accessible in chromatin. The lack of CBP in Huntington's affected neurons may lead to transcriptional repression of key genes that in turn leads to neurodegeneration (Ref.3). Huntingtin’s association with REST (Repressor Element-1 Transcription Factor) also enhances transcriptional derepression of NRSE (Neuron Restrictive Silencer Element)-regulated target genes. These genes encode a broad range of proteins involved in neuronal function and development and include neurotransmitter receptors; neurotrophins, such as BDNF (Brain-Derived Neurotrophic Factor); synaptic vesicle proteins; cytoskeletal proteins; growth factors; and ion channels. Wild-type Huntingtin, but not expanded repeat mutant Huntingtin, stimulates transcription of the gene encoding BDNF. The BDNF promoter contains an NRSE. REST/NRSF (Neuron-Restrictive Silencer Factor) binds mammalian Sin3A (Sin3 Homolog-A Transcriptional Regulator) and HDAC2 (Histone Deacetylase-2) and requires Histone Deacetylase activity to repress neuronal gene transcription. CoREST functions as an additional corepressor for REST. Wild-type Huntingtin was found to interact with REST/NRSF and, furthermore, be required to keep REST/NRSF in the cytosol, thereby preventing the formation of the corepressor complex and allowing neuronal expression of the gene. In Huntington disease, neuronal genes with NRSEs, such as BDNF and Penk (encoding Proenkephalin), are expressed at lower levels owing to the presence of mutant Huntingtin instead of two intact copies of wild-type Huntingtin. Without normal levels of wild-type Huntingtin available to bind REST/NRSF in the cytosol, more of this silencing factor can enter the nucleus to repress transcription (Ref.4). Mutant Huntingtin also disrupts transcriptional activation by SP1 and TAFII130 in HD. The transcription factor SP1 binds to DNA elements called GC boxes in cellular promoters. A specific protein-protein interaction between the glutamine-rich regions of SP1 and the TAFII130 subunit of TFIID (Transcription Factor-IID) is required for recruitment of the general transcriptional machinery, which includes transcription factors TFIIA (Transcription Factor-IIA), TFIIB (Transcription Factor-IIB), TFIID, TFIIE (Transcription Factor-IIE), TFIIF (Transcription Factor-IIF), and TFIIH (Transcription Factor-IIH). This glutamine interface serves to bridge SP1 to the machinery required to recruit RNA Pol-II (Polymerase (RNA) II (DNA directed)). Once correctly targeted to the dopamine D2 receptor gene, RNA Pol-II initiates transcription of an mRNA copy of this gene. In HD, the glutamine expansion in Huntingtin disrupts transcriptional activation by SP1 and TAFII130. The mutant Htt associates with SP1 and TAFII130, preventing SP1 from binding to the GC box, and ultimately disrupting the ability of SP1 and TAFII130 to interact. Without proper targeting by the general transcriptional machinery, RNA Pol-II cannot properly locate the Dopamine D2 receptor promoter region and the gene cannot be transcribed. The deregulated expression of this gene, as well as of many others, may be an early step in the neurodegenerative process-taking place in the HD brain (Ref.5). Under normal circumstances, Htt also heterodimerize with HIP1 (Huntingtin Interacting Protein-1). HIP1 directly associates with Clathrin heavy chain as well as Alpha-Adaptin A and C. These proteins are major components of Clt (Clathrin)-coated vesicles and are important for receptor-mediated endocytosis at the Plasma membrane. The HIP1-Huntingtin heterodimer is involved in exocytosis of neurotransmitter. Increase in the size of the polyglutamine repeat reduces the interaction with Huntingtin and HIP1. Free HIP1 can bind to HIPPI (HIP1 Protein Interactor). The HIP1/HIPPI heterodimer can recruit and activate Procaspase8, leading to apoptosis of the neuron (Ref.6). Thus it is found that Huntingtin has an important constitutive role in neurons during brain development, that heterogeneity in neuronal expression of the protein is developmentally regulated, and that the intraneuronal distribution of Huntingtin increases in parallel with neuronal maturation. The presence of mutant Huntingtin in the immature HD brain raises the possibility that neurons may be affected during brain development and possibly in the postnatal period when vulnerability to excitotoxic injury is at its peak (Ref.1 & 7). References: 1. Qin ZH, Gu ZL. Huntingtin processing in pathogenesis of Huntington disease. Acta Pharmacol. Sin Oct;25(10): PubMed ID: 2. Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR, Aronin N, DiFiglia M. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J. Biol. Chem Mar 1;277(9): PubMed ID: 3. Nucifora FC Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by Huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science Mar 23;291(5512): PubMed ID: 4. Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet Sep;35(1):76-83. PubMed ID: 5. Freiman RN, Tjian R. Neurodegeneration. A glutamine-rich trail leads to transcription factors. Science Jun 21;296(5576): PubMed ID: 6. Oravecz-Wilson KI, Kiel MJ, Li L, Rao DS, Saint-Dic D, Kumar PD, Provot MM, Hankenson KD, Reddy VN, Lieberman AP, Morrison SJ, Ross TS. Huntingtin Interacting Protein 1 mutations lead to abnormal hematopoiesis, spinal defects and cataracts. Hum. Mol. Genet Apr 15;13(8): PubMed ID: 7. Li SH, Li XJ. Huntingtin and its role in neuronal degeneration. Neuroscientist Oct;10(5): PubMed ID: Neurotranszimittert tartalmazó vezikulum mozgatása a mikrotubulusok mentén: Huntingtin – huntingtin-related-protein (HAP) – dynactin - dynein
 of a polyglutamine repeats in Huntingtin, a 350-kDa protein of unknown function. The polyglutamine repeat is localized in the N-terminal region of Huntingtin and is encoded by exon1 of the HD or Htt (Huntingtin) gene. Huntington disease is characterized by uncontrolled movements, personality changes, and dementia and causes the death of patients within 10–20 years after the appearance of the first symptoms. Huntingtin is highly expressed in the brain, and particularly enriched in Cerebral Cortex and Striatum. It is a cytoplasmic protein that is essential during development for Gastrulation and Neurogenesis, and it is important for Neuronal survival in the adult. Wild-type Huntingtin is anti-apoptotic in neurons in the Central Nervous System. Wild-type Huntingtin also reduces the toxicity of mutant Huntingtin in vivo. Huntingtin is also involved in vesicle trafficking in the secretory and endocytic pathways (Ref.1). Huntingtin shuttles between the nucleus and the cytoplasm. Full-length Huntingtin is predominantly distributed in the cytoplasm, whereas N-terminal fragments of Huntingtin with expanded polyglutamine tracts accumulate in the nucleus. Alternate Huntingtin conformations, are associated with cytoplasmic speckles and nuclear speckles or with perinuclear membranes and nucleoli. Moreover, its front end can associate with dozens of signal transduction proteins. This cargo includes members of transcription complexes [basal transcription factors (for example, SP1, TAFII130 (TBP-Associated Factor, RNA Polymerase II)), transcriptional co-repressors and co-activators (for example, NCOR1 (Nuclear Receptor Co-Repressor-1), Sin3A, CTBP (C-Terminal-Binding Protein), REST (RE1-Silencing Transcription Factor), p53), spliceosome and polyadenylation factors (for example, FBP11, symplekin)], a ubiquitin protein ligase (E2-25 kD) that regulates transcription factor turnover, effectors [receptors (for example, NMDAR (N-methyl-D-aspartate calcium channel receptors), EGFR (Epidermal Growth Factor Receptor)), Kinase (MLK2 (Mixed Lineage Kinase-2)), and Phosphatase (SHP2)], and adaptor proteins that localize signaling complexes (for example, GRB2 (Growth Factor Receptor-Bound Protein-2), PACSIN1 (Protein Kinase-C and Casein Kinase Substrate in Neurons-1), PSD95 (Postsynaptic Density-95), HAP1 (Huntingtin’s associated protein-1). Huntington disease pathogenesis can be viewed as a cascade of events that is first triggered by mutant Huntingtin through an abnormal interaction with an as-yet unknown cellular constituent. Once triggered, this cascade broadens with time, leading eventually to neuronal cell death (Ref.2). The Huntingtin’s expanded polyglutamine alters protein conformation, resulting in aberrant protein interactions, including interactions of the expanded polyglutamine with cellular proteins containing short polyglutamine stretches. CBP (CREB Binding Protein) is a co activator for CREB (cAMP Response Element-Binding Protein)-mediated transcription and contains an 18-glutamine stretch. CREB-mediated gene transcription promotes cell survival, and CBP is a major mediator of survival signals in mature neurons. CBP is a large protein with multiple domains and in addition to interacting with several different transcription factors also has a Histone Acetylase enzyme activity. The expanded polyglutamine in HD can interact with the short glutamine repeat in CBP, interfering with CBP function, causing transcriptional abnormalities, and leading to cellular toxicity. Mutant Huntingtin binds to CBP, drawing it into insoluble protein aggregates. Mutant Huntingtin also interacts directly with the acetyltransferase domain of CBP, blocking this activity. Activation of CBP acetyltransferase activity by transcriptional regulators results in the acetylation of histones in the promoter and enhancer regions of active genes, contributing to transcriptional activation by making these genes more accessible in chromatin. The lack of CBP in Huntington s affected neurons may lead to transcriptional repression of key genes that in turn leads to neurodegeneration (Ref.3). Huntingtin’s association with REST (Repressor Element-1 Transcription Factor) also enhances transcriptional derepression of NRSE (Neuron Restrictive Silencer Element)-regulated target genes. These genes encode a broad range of proteins involved in neuronal function and development and include neurotransmitter receptors; neurotrophins, such as BDNF (Brain-Derived Neurotrophic Factor); synaptic vesicle proteins; cytoskeletal proteins; growth factors; and ion channels. Wild-type Huntingtin, but not expanded repeat mutant Huntingtin, stimulates transcription of the gene encoding BDNF. The BDNF promoter contains an NRSE. REST/NRSF (Neuron-Restrictive Silencer Factor) binds mammalian Sin3A (Sin3 Homolog-A Transcriptional Regulator) and HDAC2 (Histone Deacetylase-2) and requires Histone Deacetylase activity to repress neuronal gene transcription. CoREST functions as an additional corepressor for REST. Wild-type Huntingtin was found to interact with REST/NRSF and, furthermore, be required to keep REST/NRSF in the cytosol, thereby preventing the formation of the corepressor complex and allowing neuronal expression of the gene. In Huntington disease, neuronal genes with NRSEs, such as BDNF and Penk (encoding Proenkephalin), are expressed at lower levels owing to the presence of mutant Huntingtin instead of two intact copies of wild-type Huntingtin. Without normal levels of wild-type Huntingtin available to bind REST/NRSF in the cytosol, more of this silencing factor can enter the nucleus to repress transcription (Ref.4). Mutant Huntingtin also disrupts transcriptional activation by SP1 and TAFII130 in HD. The transcription factor SP1 binds to DNA elements called GC boxes in cellular promoters. A specific protein-protein interaction between the glutamine-rich regions of SP1 and the TAFII130 subunit of TFIID (Transcription Factor-IID) is required for recruitment of the general transcriptional machinery, which includes transcription factors TFIIA (Transcription Factor-IIA), TFIIB (Transcription Factor-IIB), TFIID, TFIIE (Transcription Factor-IIE), TFIIF (Transcription Factor-IIF), and TFIIH (Transcription Factor-IIH). This glutamine interface serves to bridge SP1 to the machinery required to recruit RNA Pol-II (Polymerase (RNA) II (DNA directed)). Once correctly targeted to the dopamine D2 receptor gene, RNA Pol-II initiates transcription of an mRNA copy of this gene. In HD, the glutamine expansion in Huntingtin disrupts transcriptional activation by SP1 and TAFII130. The mutant Htt associates with SP1 and TAFII130, preventing SP1 from binding to the GC box, and ultimately disrupting the ability of SP1 and TAFII130 to interact. Without proper targeting by the general transcriptional machinery, RNA Pol-II cannot properly locate the Dopamine D2 receptor promoter region and the gene cannot be transcribed. The deregulated expression of this gene, as well as of many others, may be an early step in the neurodegenerative process-taking place in the HD brain (Ref.5). Under normal circumstances, Htt also heterodimerize with HIP1 (Huntingtin Interacting Protein-1). HIP1 directly associates with Clathrin heavy chain as well as Alpha-Adaptin A and C. These proteins are major components of Clt (Clathrin)-coated vesicles and are important for receptor-mediated endocytosis at the Plasma membrane. The HIP1-Huntingtin heterodimer is involved in exocytosis of neurotransmitter. Increase in the size of the polyglutamine repeat reduces the interaction with Huntingtin and HIP1. Free HIP1 can bind to HIPPI (HIP1 Protein Interactor). The HIP1/HIPPI heterodimer can recruit and activate Procaspase8, leading to apoptosis of the neuron (Ref.6). Thus it is found that Huntingtin has an important constitutive role in neurons during brain development, that heterogeneity in neuronal expression of the protein is developmentally regulated, and that the intraneuronal distribution of Huntingtin increases in parallel with neuronal maturation. The presence of mutant Huntingtin in the immature HD brain raises the possibility that neurons may be affected during brain development and possibly in the postnatal period when vulnerability to excitotoxic injury is at its peak (Ref.1 & 7). References: 1. Qin ZH, Gu ZL. Huntingtin processing in pathogenesis of Huntington disease. Acta Pharmacol. Sin Oct;25(10): PubMed ID: Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR, Aronin N, DiFiglia M. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J. Biol. Chem Mar 1;277(9): PubMed ID: Nucifora FC Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by Huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science Mar 23;291(5512): PubMed ID: Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet Sep;35(1): PubMed ID: Freiman RN, Tjian R. Neurodegeneration. A glutamine-rich trail leads to transcription factors. Science Jun 21;296(5576): PubMed ID: Oravecz-Wilson KI, Kiel MJ, Li L, Rao DS, Saint-Dic D, Kumar PD, Provot MM, Hankenson KD, Reddy VN, Lieberman AP, Morrison SJ, Ross TS. Huntingtin Interacting Protein 1 mutations lead to abnormal hematopoiesis, spinal defects and cataracts. Hum. Mol. Genet Apr 15;13(8): PubMed ID: Li SH, Li XJ. Huntingtin and its role in neuronal degeneration. Neuroscientist Oct;10(5): PubMed ID: Neurotranszimittert tartalmazó vezikulum mozgatása. a mikrotubulusok mentén: Huntingtin – huntingtin-related-protein (HAP) – dynactin - dynein.")
52
Összefüggés a ‘CAG’ repeat-szám és a megjelenés
időpontja köztött

53
George Huntington ( ) Nagyapja és apja vidéki orvosok voltak, az ő diagnosztikus adataik is segítették a betegség leírásában A tünetek leírása 1872 Medical and Surgical Reporter of Philadelphia chorea = mániás tánc

54
Trinukleotid-repeat CCG

55
Anticipáció – Trinukleotid repeat
Az egymást követő generációkban a betegség egyre súlyosabb formában (egyre korábban) jelentkezik
 jelentkezik.")
56
Autoszómális Recesszív A tüneteket mutató személy
szülei rendszerint tünetmentesek (Aa) Mindkét nembeli érintett lehet Ffi és nő egyaránt átörökít Gyakori a szülők vérrokonsága Beteg születésének kockázata 25% (Aa x Aa) Horizontális családfa
 Mindkét nembeli érintett lehet. Ffi és nő egyaránt átörökít. Gyakori a szülők vérrokonsága. Beteg születésének kockázata. 25% (Aa x Aa) Horizontális családfa.")
57
Autoszómális Recesszív A tüneteket mutató személy
szülei rendszerint tünetmentesek (Aa) Mindkét nembeli érintett lehet Ffi és nő egyaránt átörökít Gyakori a szülők vérrokonsága Beteg születésének kockázata 25% (Aa x Aa) Horizontális családfa
 Mindkét nembeli érintett lehet. Ffi és nő egyaránt átörökít. Gyakori a szülők vérrokonsága. Beteg születésének kockázata. 25% (Aa x Aa) Horizontális családfa.")
58
Recesszív autoszómális
1700 ismert emberi recesszív jelleg Több mint 15% enzim-hiba - enzimopathia (ld. fenilalanin hidroxiláz, hexóz aminidáz) Hemoglobin mutációk Multiplex allélizmus – a jelleg kialakulásáért egy adott gén számos mutációja lehet felelős Komplex heterozigóta – aki egy adott gén két eltérő, mutáns allélját hordozza Pl. Cisztás fibrózis – 850 különböző mutáció
 Hemoglobin mutációk. Multiplex allélizmus – a jelleg kialakulásáért egy adott gén számos mutációja lehet felelős. Komplex heterozigóta – aki egy adott gén két eltérő, mutáns allélját hordozza. Pl. Cisztás fibrózis – 850 különböző mutáció.")
59
Recesszív betegségek gyakorisága
Egyes etnikai csoportokban jelentősen eltérő OK: heterozigóták reprodukciós előnye a homozigótákkal szemben KÖRNYEZETI TÉNYEZŐK Heterozigóták szelekciós előnye Egyéb ok: egyes etnika csoportokban irányított párválasztás

60
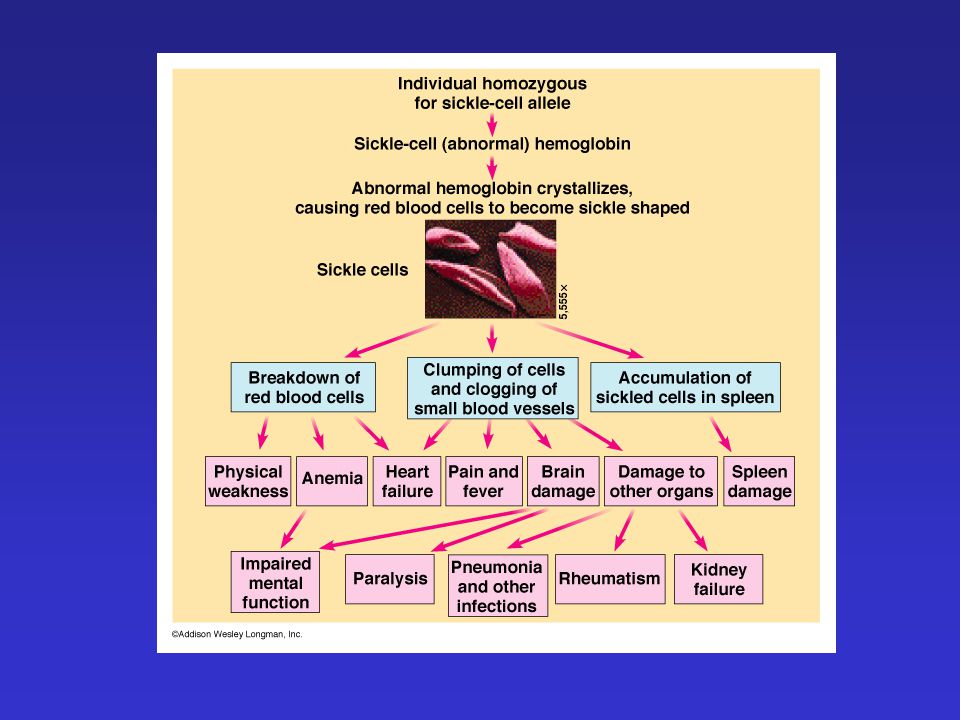
Sarlósejtes anémia Autoszomális-Rec.. Hemoglobinopátia
HBB gén – 11 krsz. b-globin lánc mutációja HbS variáns

61
Hemoglobin szerkezeti változása
HbA HbS

62
A HbS rendellenes szerkezete miatt:
A vvt.-ek membránja sérül Periférián, (vese/lép) nagy számban eliminálódnak O2 szállítás zavart
 nagy számban eliminálódnak. O2 szállítás zavart.")
64
Malária terjedése Plasmodium falciparum Plasmodium vivax

65
Heterozigóták szelekciós előnye:
Malária Sarlósejtes anémia

66
Heterozigóták szelekciós előnye:
Malária Sarlósejtes anémia DE: a környezet változásával az arány is csökken. Pl. Cipruson a thalasszémia gyakorisága csökken

67
b-Thalasszémia (thalassa = tenger)
")
68
b-Thalasszémia (thalassa = tenger) hemoglobin b lánc mutációi
vvt. rövid élettartam O2 szállító kapacitás csökken

69
Cisztás fibrózis

70
Cisztás fibrózis Szelekciós előny: Kolera nagy Cl- veszteséggel járó
vezető tünete a tubuláris szerkezető szervek elzáródása érintett szervek: tüdő, hasnyálmirigy, ivarszervek jellegzetes tünet: verejték Cl- conc. süketség Szelekciós előny: Kolera nagy Cl- veszteséggel járó betegség esetén - Aa heterozigóták (aa – CF beteg, AA – kolera beteg, Aa CF – nem veszít Cl- ionokat)
")
71
Cisztás fibrózis az F508 (Phe) deléciója
a del. egy ABC transzportert érint Cl- ionok sejtből történő eltávolításában van szerepe

72
Cisztás fibrózis klorid-csatorna mutációja 1/25 gyakoriság 850 mutáció

73
lerakódások agyban (LM és TEM)
Tay-Sachs betegség GM2 gangliozid lebontási zavara HEAXA gén b-hexózaminidáz enzim érintett (idegsejtek lizoszómái) Idegrendszeri érintettség: - paralízis - demencia - vakság - korai halál GM2 gangliozid lerakódások agyban (LM és TEM)
 Idegrendszeri érintettség: - paralízis. - demencia. - vakság. - korai halál. GM2 gangliozid. lerakódások agyban (LM és TEM)")
74
Tay-Sachs betegség Lysosome membrane phospholipid GM2 GM2 activator
Hex A

75
Tay-Sachs betegség HA-a alegységen a Man foszforiláció elmarad
Az enzym nem transzportálódik lizoszómába

76
Tay-Sachs betegség Környezeti hatás: Hordozók gyakorisága: 1:300
15q23-q24 Hordozók gyakorisága: 1:300 Askenázi zsidó populációban: 1:30 Környezeti hatás: Galíciában - összezárva, rossz higiénés viszonyok között éltek - TBC miatti magas halálozás – AA homozigóták - Tey-Sachs miatti halálozás – aa homozigóták Aa heterozigóták – reprodukciós előnnyel bírtak

77
Egyéb recesszív autoszómálisan öröklődő betegségek

78
Egyéb recesszív autoszómálisan
öröklődő betegségek 12 Fenilketonúria - fenilalanin hidroxiláz hiányos működése - mentális retardáció - lassú fejlődés 11, 15, 9, X Albinizmus - Tyr -> melanin képzés zavara - Enzim def.: tirozináz enzim zavara bőr, szem érintett több forma ismert Galaktozémia

79
Albino és normal kenguruk
Albino Barking Deer Albino rák Albino prérifarkas

80
Etnikai eltérések hatása egyes öröklődő betegségek megjelenésére

Hasonló előadás







 Bihari Péter.>")




 kialakulása Genetikai, Sejt- és Immunbiológiai Intézet Falus András.>")








