Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
Genetikai vizsgálatok jelentősége
Dr. Tímár László

2
Preventív medicina része
Genetikai tanácsadás Preventív medicina része Feladatai 1. Családtervezők segítése 2. Hatékony orvosi ellátás előmozdítása. 3. Ártalmak megelőzése

3
Utódvállaláskor jelentkező Specifikus veszély Általános veszély

4
Genetikai tanácsadás folyamata
1. Előzetes tájékozódás, családfa felvétel 2. A pontos kóroki diagnózis megállapítása. 3. A specifikus veszély mértékének meghatározása. 4. Tanácsadás Fontos: A döntés mindig a családtervezők joga, feladata és felelőssége!

5
A specifikus veszély megállapításának 10 szempontja
1. Súlyosság 2. Kezelhetőség 3. Méhen belüli felismerhetőség 4. Genetikai kockázat 5. Maternális veszély 6. Teratogén veszély 7. Postnatalis veszély 8. Psychológiai effectus 9. Meglévő gyermekek 10. Család körülményei

6
1. Súlyosság Reprod. életkor előtt halálos Önálló társadalmi tevékenységre képtelen 2. Kezelhetőség Specifikus terápia lehetősége 3. Prenatalis diagnosztika lehetősége CF; SMA; DMD; stb. 4. Genetikai kockázat I. Monolocusos AD, AR, XR II. Kromoszómális III. Polygénes - Multifaktoriális

7
A proximalis spinalis izomatrophia (SMA) etiológiája 1.
gerincvelői alfa-motoneuronok fokozatos degenerációja jellegzetes klinikai tünetek: hypotonia, progresszív szimmetrikus izomgyengeség végtagok, majd a törzsizomzat bénulása histopathológia: atrophiás (1. vagy 2. típusú) valamint csoportosult hypertrophiás (1. típusú) rostok (kollaterális innerváció) EMG: denervációs jelek, óriás potenciálok
 valamint csoportosult hypertrophiás (1. típusú) rostok (kollaterális innerváció) EMG: denervációs jelek, óriás potenciálok.")
8
A proximalis spinalis izomatrophia (SMA) etiológiája 2.
5 különböző súlyosságú fenotípus csoport congenitalis forma, SMA 0: halál <4 hó Werdnig-Hoffmann kór (SMA I): kezdet <6 hó, halál <18 hó, nem ül Fried-Emery (SMA II): kezdet <18 hó, halál >2 év, nem jár Kugelberg-Welander (SMA III): kezdet>18 hó, halál felnőttkorban, jár felnőttkori forma, SMA IV: kezdet >30 év, normális élettartam, genetikailag heterogén
: kezdet <6 hó, halál <18 hó, nem ül. Fried-Emery (SMA II): kezdet <18 hó, halál >2 év, nem jár. Kugelberg-Welander (SMA III): kezdet>18 hó, halál felnőttkorban, jár. felnőttkori forma, SMA IV: kezdet >30 év, normális élettartam, genetikailag heterogén.")
9
Az SMA genetikai háttere
gyakoriság: 1/6000, hordozók 1/35 5q13 lókuszban komplex genetikai régió (500 kb), ismétlődő, invertált szekvenciákkal, nagyméretű deléciónak kitéve SMN (survival of motoneuron) gén azonosítása egyéb modifikáló gének (NAIP, p44, H4F5, TFNR- transzkripciós faktor, HRAD17-sejtciklus szabályozó gén) tel cen p44c(BTF2) NAIPY SMNc SMNt NAIP5 p44t(BTF2) XS2G3 C212 C272
, ismétlődő, invertált szekvenciákkal, nagyméretű deléciónak kitéve. SMN (survival of motoneuron) gén azonosítása. egyéb modifikáló gének (NAIP, p44, H4F5, TFNR- transzkripciós faktor, HRAD17-sejtciklus szabályozó gén) tel. cen. p44c(BTF2) NAIPY. SMNc. SMNt. NAIP5. p44t(BTF2) XS2G3. C212. C272.")
10
SMA Genetikai vizsgálat
A beteg genotípusa a kiindulási alap - a beteg DNS mintája nélkülözhetetlen - DNS bankok létrehozása PCR technika - vércsepp (Guthrie kártya) analízisre is.
 analízisre is.")
11
SMA Vizsgálati módszerek
haplotipizálás a gént körülvevő DNS markerekkel direkt mutáció analízis - deléció azonosítás az SMN gén 7. és 8. exonjában

12
Kromoszóma vizsgálat indikációi
1 – MCA-val sújtott páciens 2 – Craniofacialis dysmorphia + Ment. ret. 3 – Minor – major malformatio + kp. Idegrendszeri dysfunkció 4 – Jól ismert kromoszóma szindróma karakterisztikus tünetei 5 - Nemi szervi fejl. rend. 6 – Habituális abortusz 7 – Kiegyensúlyozott kromoszóma transzlok. carrier

15
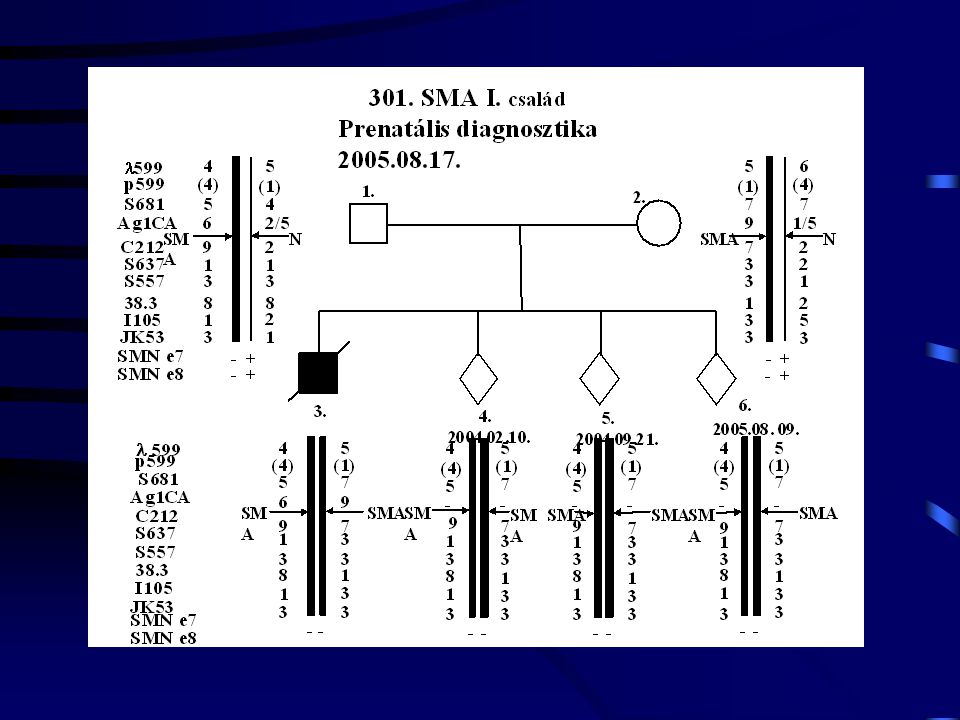
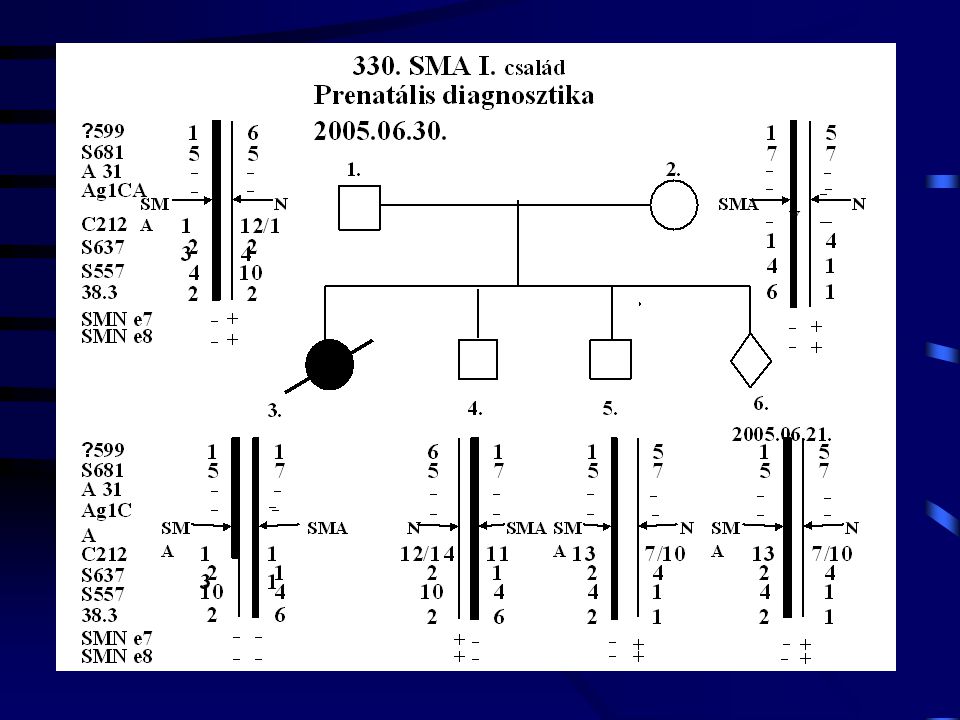
3 C 1988- UM: 1800 g. Exit: 4000 g. SMA-III. 2900 g. SMA

16
C 1987- 3350 g. 2950 g. DMD DMD

17
SMA (Non pat. + cons.) 1995. 04. 10. 2950 g. Exit: 1995. 09. 12. SMA-I
1100 g. Exit: 3200 g. SMA (Non pat. + cons.)
")
18
Általános jellemzők, klinikai tünetek
Előfordulási gyakoriság - DMD 1:3500, BMD 1:30000 X kromoszómához kötött recesszív öröklődésmenet A betegség kezdete 2 éves korra tehető, fokozatos progresszióval Az elhalálozás ideje éves korban A Becker típus esetén enyhébbek a tünetek, nincs korai halál

19
A betegség genetikai háttere, pathomechanizmusa
A dystrophin gén (Xp21) Mb méretű, 79 exont tartalmaz és 14 kb méretű mRNS-t kódol A géntermék a dystrophin fehérje (427 kDa), amely sarcolemmális lokalizációt mutat a dystrophin- asszociált glycoprotein komplex részeként A DMD esetén a dystrophin teljes hiánya, BMD esetén csökkent mennyisége mutatható ki
 2.5 Mb méretű, 79 exont tartalmaz és 14 kb méretű mRNS-t kódol. A géntermék a dystrophin fehérje (427 kDa), amely sarcolemmális lokalizációt mutat a dystrophin- asszociált glycoprotein komplex részeként. A DMD esetén a dystrophin teljes hiánya, BMD esetén csökkent mennyisége mutatható ki.")
20
C DMD DMD

21
Genetikai háttér NF 1 AD Penetrancia: 100%
Expressivitas:rendkívül variábilis Prevalencia: 1: 3000 Páciensek fele familiáris, felénél új mutáció Új NF 1 mutációk 80%-a paternalis eredetű Mutációs ráta: 1: Homozygota NF 1 beteget még nem írtak le Ok: haploinsufficiencia Somaticus mozaikosság Germinális mozaikosság

22
Genetikai háttér NF 1: A variabilis expresivitás lehetséges magyarázatai: Allél heterogenitás Somaticus mozaikosság Modifikáló gének Környezeti faktorok

23
Genetikai háttér NF 2: AD Penetrancia: 95% Expressivitás: variabilis
Prevalencia: 1 : Az esetek 50-75%-a új mutáció következménye

24
Molekuláris genetikai háttér
NF 1 Locus: 17q11.2 NF 1 gént 1990-ben azonosították Nagy gén – 350 kilobasis, 60 exon Az NF 1 gén által kódolt fehérje a neurofibromin, mely 2818 aminósavból áll és 327 kDA súlyú. NF 1 tumor suppressor gén. Nerofibromin domain homológja a GTPase aktiváló fehérje (GAP) családnak, szabályozza a p21 ras proto- oncogen aktivitást. A neurofibromin funkciójának hiánya kontrollálatlan sejtnövekedéshez, tumor kialakuláshoz vezethet.
 családnak, szabályozza a p21 ras proto- oncogen aktivitást. A neurofibromin funkciójának hiánya kontrollálatlan sejtnövekedéshez, tumor kialakuláshoz vezethet.")
25
Genotypus-phenotypus összefüggés
NF 1: Nincs összefüggés a mutáció tipusa és a betegség klinikai súlyossága között, kivéve a teljes génre kiterjedő nagy deléciót ! Ez az NF 1 betegek 4-7%-nál fordul elő, igen súlyos tünetekkel jár, korai kezdetű. Craniofaciális dysmorphiás tünetek és mentalis retardatió észlelhető. Kimutatás: FISH

26
Genotypus-phenotypus összefűggés
NF 2: 1. Súlyos tünetekkel érintett pácienseknél deléciót, insertiót vagy nonsens mutációt észleltek 2. Enyhe tünetekkel érintett pácienseknél missense mutációt észleltek

27
Molekuláris genetikai háttér
NF 2 Locus: 22q12.2 NF 2 gént 1993-ben azonosították Fehérje termék: merlin, melynek funkciója potosan nem tisztázott NF 2 tumor suppressor gén

28
Molekuláris genetikai vizsgálati lehetőségek
PTT – Protein Truncation Test -RNS izolálás - reverse transcriptio – NF 1 complementer DNS-fragment – neurofibromin szintézis in vitro – polyacrilamid gél elektroforézis – szeparálás – súly változása alapján. Alacsony a sensitivitása: a biztosan NF betegek 70%-ban tudja csak azonosítani a mutációt. Nem mutatja ki a nagy deléciókat a missence mutációkat és a mozaikosságot sem. Direct test a specifikus mutáció kimutatására Familiáris NF esetekben ha legalább 2 vagy több érintett személy van akkor linkage analysis is végezhető intragenikus microsatellita NF 1 markerekkel

29
Fragilis X betegség Klinikai tünetek
X kromoszómához kötött értelmi fogyatékosság férfiak: IQ = 37-70, autizmus, hiperaktivitás, jellegzetes arcforma, nagy elálló fülek, makroorchidizmus, kötöszöveti lazaság nõk: hordozók 30%-ában mentális tünetek, enyhébb értelmi fogyatékosság, kognitív és pszichés zavarok Prevalencia férfiak: / nõk: / 1000 hazai becslések (Czeizel 1997): minden újszülöttben Þ kb. 70 új eset évente Þ 45 fiú+25 lány minden népességben elõfordul
: minden újszülöttben Þ kb. 70 új eset évente Þ 45 fiú+25 lány. minden népességben elõfordul.")
30
KLINIKAI TÜNETEK Mérsékelt – változó súlyosságú mentális retardatió IQ: 35 – 55. Beszédfejlődési zavar- gyakori szóismétlés. Autizmus. Hiperkinetikus hiperaktivitás. Dekoncentráltság. Figyelemzavar. Emocionális instabilitás. Néha saját kéz harapdálása. Craniofacialis dysmorphia: Macrocephalia,kiemelkedő homlok, előreugró,megvastagodott mandibula,nagy elálló fülek,hosszúkás arcforma. Macroorchidismus (megalotestis) Kötőszöveti lazaság. Laza izületek főleg az ujjakon.
 Kötőszöveti lazaság. Laza izületek főleg az ujjakon.")
31
KÉT FRAXA BETEG FIVÉR

32
Genetikai háttér Cytogenetika - Xq27.3 indukált töréspontok detektálása (FRAXA lókusz) - nem megbízható, hordozók kimutatására és prenatális vizsgálatra nem alkalmas Molekuláris genetika - FMR-1 gén felfedezése (Verkerk 1991) - CpG sziget Þ (CGG)n ismétlõdések kóros expanziója a gén 5’ végén, 1. exonban
 - nem megbízható, hordozók kimutatására és prenatális vizsgálatra nem alkalmas. Molekuláris genetika - FMR-1 gén felfedezése (Verkerk 1991) - CpG sziget Þ (CGG)n ismétlõdések kóros expanziója a gén 5’ végén, 1. exonban.")
33
Molekuláris háttér I. 1. normál gén: (CGG)n = 5-50, mitotikusan és meiotikusan is stabil 2. premutáció: (CGG)n = , nincs metilálva, mitotikusan stabil, meiotikusan instabil 3. teljes mutáció: (CGG)n > 200, teljesen metilált, mitotikusan és meiotikusan is instabil 4. jelentős expanzió generációk között, ha nõ örökíti tovább premutáció - teljes mutáció átmenet a petesejt meiózisa alatt vagy postzygotikusan
n = , nincs metilálva, mitotikusan stabil, meiotikusan instabil. 3. teljes mutáció: (CGG)n > 200, teljesen metilált, mitotikusan és meiotikusan is instabil. 4. jelentős expanzió generációk között, ha nõ örökíti tovább. premutáció - teljes mutáció átmenet a petesejt meiózisa alatt vagy postzygotikusan.")
34
Molekuláris háttér II. 5. metiláció mozaikossága különbözõ szomatikus sejtekben 6. prenatális vizsgálatok Þ speciális óvatosság chorion és magzati szövetek alulmetilált állapota miatt (13. hétig) 7. teljes mutációs nőknek csak 50-70%-ában klinikai tünetek (enyhébbek) Þ csökkent penetranciát random X inaktiváció szabályozza Betegség súlyossága korrellál a (CGG)n mértékével és a metiláció fokával X-hez kötött domináns trinukleotida expanziós betegség anticipációval, nőkben nem teljes penetranciával
 7. teljes mutációs nőknek csak 50-70%-ában klinikai tünetek (enyhébbek) Þ csökkent penetranciát random X inaktiváció szabályozza. Betegség súlyossága korrellál a (CGG)n mértékével és a metiláció fokával. X-hez kötött domináns trinukleotida expanziós betegség anticipációval, nőkben nem teljes penetranciával.")
35
Anticipáció Egymást követő generációkban egyre súlyosabb a betegség és egyre koraibb a manifesztáció Variábilitás speciális formája Trinukleotida expanzióval jól magyarázható repeat szám növekedésével jól korrelál. Pl.: FraX, Dystrophia myotonica, Huntington

36
Nem teljes penetrancia A felmenő ági rokonok célzott vizsgálata!
Adott genotípus penetranciája = phenotípusos tünetek megjelenésének valószínűsége AD öröklődésmenetet bonyolítja GT problémája. Több tényező szabályozza Egyéb genetikai faktorok és környezet hatása érvényesülhet Pl.: CMT Fontos: A felmenő ági rokonok célzott vizsgálata!

Hasonló előadás
















>")


