Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
Bioinformatika - Proteomika
Medzihradszky (Fölkl) Katalin Adjunct Professor of Pharmaceutical Chemistry, University of California San Francisco és Proteomikai Kutatócsoport, SzBK Csoportvezető, tudományos főmunkatárs vagy
 Katalin. Adjunct Professor of Pharmaceutical Chemistry, University of California San Francisco. és. Proteomikai Kutatócsoport, SzBK. Csoportvezető, tudományos főmunkatárs. vagy")
2
Bioinformatika úgy általában
adatbázisok felépítése ezen adathalmazok statisztikai analízise, matematikai értelmezése szabályszerűségek meghatározása szabályok hasznosítása – predikciók pl. funkcióra, rokonságra, eredetre, szerkezetre stb.

3
Bioinformatika úgy általában
„összehordott” adatokból építkezünk többnyire elmélet eleinte a gyakorlati hasznosítás majd a végén jön persze ez visszahat az elméletre

4
Mennyire megbízhatóak az adatok?
Mit lehet kiaknázni? genomiális adatbázisok fehérje szekvencia adatbázisok fehérje 3D szerkezeti adatbázisok stb, stb Meghatározó faktor: Mennyire megbízhatóak az adatok?

5
A sejt fehérjetartalmának kvalitatív és kvantitatív jellemzése
Proteomika A sejt fehérjetartalmának kvalitatív és kvantitatív jellemzése Mi van jelen? Mennyi? Milyen formában? Lokalizáció? Kölcsönhatások? Partnerek?

6
A proteomika különleges jellege
nagyon is gyakorlati tudomány adatokat produkálunk, amit informatika segítségével adatbázisokból értelmezünk bővítjük/építjük az adatbázisokat, ill. új adatbázisokat hozunk létre új bioinformatikai eszközöket fejlesztünk ki az eredmények alapján

7
Problémák állandó dinamikus változások hatalmas mennyiségi különbségek
igen eltérő fizikai tulajdonságok/ vagy csak árnyalatnyi különbségek – ugyanannak a génnek a termékeire

8
Még mindig „problémák”
Poszt-transzlációs módosítások Idő-, faj-, állapot-, lokalizáció- stb. -függő permanens vs. dinamikus teljes vs. részleges jelentős vs. csekély méretváltozás hidrofil hidrofób Nem árt érteni a biológiához!

9
Láthatóvá tenni a komplexitást...
Frakcionálni kell ! 1D-, 2D-, multidimenziós módszerek

10
Mi mindent kell meggondolni?
Mennyire univerzális a módszer? Felbontása? Dinamikus tartománya? Követhetősége? Reprodukálhatósága? Kvantitatív-e?

11
1D-SDS-PAGE * Minden belemegy a gélbe, de kicsi a felbontás.
* Várhatóan fehérje-elegyeket kell analizálni.

12
2D-elfo 1. dimenzió: izoelektromos fókuszálás → pI szerint - pH 3-10
savas vagy bázikus fehérjék NEM fókuszálódnak membrán-fehérjék kicsapódnak – ionos detergens nem használható 2. dimenzió: SDS-PAGE

13
Több példányban elkészítendő, statisztikai analízisre alkalmatos
1. spot 7. spot 8. spot 2. spot 3. spot 4. spot 5. spot 6. spot 9. spot pH: 2 proteins 2 proteins 3 proteins 3 proteins 2 proteins 1 protein 1 protein 1 protein 4 proteins Több példányban elkészítendő, statisztikai analízisre alkalmatos

14
2D-elfo másképpen 1. dimenzió: 16 BAC-PAGE (pozitív „fejű” detergens)
2. dimenzió: SDS-PAGE Minden belemegy a gélbe, de átlósan frakcionálódik – kis felbontás

15
Speciális 2D elválasztás
16-BAC S D Pros35 10/14 (71%) 33% Pros7 34/36 (94%) 63% Pros6 12/12 (100%) 42% Pros /21 (66%) 54% Pros29 19/23 (83%) 59% Pros2 13/15 (86%) 42% GC /14 (93%) 35% ProsMA5 22/44 (50%) 56% Pros25 12/15 (80%) 50% Pros3 18/24 (75%) 53% Pros26 6/15 (40%) 46% Pros5 11/12 (92%) 26% GC /23 (47%) 46% l(2) /19 (79%) 72%
 33% Pros7 34/36 (94%) 63% Pros6 12/12 (100%) 42% Pros /21 (66%) 54% Pros29 19/23 (83%) 59% Pros2 13/15 (86%) 42% GC /14 (93%) 35% ProsMA5 22/44 (50%) 56% Pros25 12/15 (80%) 50% Pros3 18/24 (75%) 53% Pros26 6/15 (40%) 46% Pros5 11/12 (92%) 26% GC /23 (47%) 46% l(2) /19 (79%) 72%")
16
Festés Commassie Brillian Blue – kvanti
Ezüst – érzékenyebb, de NEM kvanti Trükkök: funkciós csoportra specifikus festés differenciál festés

17
2D-kromatográfia 1. dimenzió: kation - ioncsere
2. dimenzió: fordított fázisú HPLC Követés: 215 nm-en → peptidkötés Nyilván semmi hidrofób nem érvényesül ilyen körülmények között... Reprodukálhatóság degradálódik az oszlop életkorával!

18
Ki is kell találni mit is válogattunk szét!
Edman szekvenálás → N-terminuson blokkolt fehérjékkel, keverékkel nem megy Western-blot → tudni kell, hogy mit keresünk, és kell jó ellenanyag

19
alapötlet Egy bizonyos fehérje adott specificitású enzimmel emésztve szekvenciájára jellemző hasítási termékeket fog produkálni Ha a valóságos emésztményt az adatbázis „in-silico” eredményeivel összevetjük azonosítani tudjuk a fehérjét Az összehasonlítás alapja csak valami biztosan megjósolható és egyértelműen meghatároztató tulajdonság lehet: TÖMEG

20
A tömegspektrometria előnyei
alkalmazható keverékekre blokkolt fehérjékre is kovalens módosítások azonosíthatóak

21
Detektálási érzékenység
Edman szekvenálás – kb. 1 pmól M MS – kb fmól - 5x M Western blot – kevesebb, mint M

22
MS-alapú Proteomika Fordított bioinformatika:
mérési adatok + adatbázisok + algoritmusok → biológiai eredmények Tudni illik valamit az analitikai módszerről

23
Mass Spectrometry 101 Ionokat mérünk
Matrix Assisted Laser Desorption Ionization Electrospray Ionization – többszörösen töltött ionok Nagy vákuumban quadrupole, Time-of-Flight, ioncsapda MS → MH+ adatok MS-MS alias CID, CAD, PSD ... → szekvencia info

24
Monoizotópos tömeg csak C12, H1, N14, O16 and S32 ← 1 C13 2 C13

25
Felbontás: m/Dm = /0.2 ~ 8000

26
Miért is kell felbontás?

27
Normál peptid, 1000-es felbontás

28
Normál peptid, 10 000-es felbontás

29
Br-Trp-tartalmú peptid, 1000-es felbontás

30
Br-Trp-tartalmú peptid, 10 000-es felbontás

31
3+ töltésű peptid, kis felbontás

32
3+ töltésű peptid, nagy felbontás
Δ = = kb. 1/3

33
Tömegmérés pontossága
0.1 Da mérési hiba egész jó 2 kDa-nál, DE katasztrófális egy kis molekulánál Relatív értéket adjunk meg! pars per million

34
Milyen pontosan tudunk mérni?
belső standarddal 20 ppm-en belül ± külső standarddal 200 ppm-en belül ± intakt fehérjékre 0.1%-on belül ± Gyenge jeleknél nem érvényesül a Gauss eloszlás!

35
Emésztési elegy MALDI-spektruma

36
Peptidek ESIMS analízise

37
Intakt fehérjék MALDI-analízise

38
Intakt fehérje ESI-analízise

39
Ha ez így megy, minek egyáltalán emésztgetni?
Miért nem mérjük az intakt fehérjéket? preparatív és interpretív akadályok

40
Minta-előkészítés Legyen a fehérje hozzáférhető a hasításra:
→ denaturálás → diszulfid-hidak bontása Emésztés, kémiai hasítás

41
Specifikus hasítások tripszin - Lys↓ Arg ↓ endoproteáz Lys-C - Lys ↓
endoproteáz Glu-C - Glu ↓ (Asp ↓) endoproteáz Asp-N - ↓Asp (↓Glu) pH kb CNBr (+75 %-os TFA) - Met ↓
 endoproteáz Asp-N - ↓Asp (↓Glu) pH kb CNBr (+75 %-os TFA) - Met ↓")
42
Egy nagyon praktikus software
Segít pl. mólsúly –számításban, „megjósolja” a hasítási vagy MS-MS termékeket ... „mindentudó”adatbázis-lekereső

47
Az analízis Egyben mérjünk vagy frakcionáljunk? MALDI vs ESIMS
LC-MALDI LC-ESIMS

48
Mennyire kellene pontosan mérni a peptideket ?
MH+ = ± 0.3 ppm 9 különböző aminosav-összetétel 36292 különböző szekvencia 3 különböző elemi összetétel Ile/Leu ThrVal/SerLeu GlyGlu/AlaAsp

49
Miért kell még szekvencia info?
Keverékek Kovalens módosítások Izoformák Splice variants És ha nincs bent az adatbázisban?

50
Hogyan tegyünk szert a szükséges információra?
Feladat: → meghatározni a komponensek tömegét → egyet fizikailag elkülöníteni → „szétverni” Műszeres megoldás: tandem tömegspektrometria, ionkapu, ioncsapda Fragmentálás: post-source decay (PSD), ütközéses aktiválás (CID/CAD), elektron-befogás (ECD)
, ütközéses aktiválás (CID/CAD), elektron-befogás (ECD)")
51
A fragmentáció módszer- és készülék-függő!
Peptid fragmentáció NH2-CH(R1)-CO-NH-CH(R2)-CO~ ~CO-NH-CH(Rn)-COOH yn-2 bn-1 yn-1 y1 b2 bi=S gyöksúly + 1 (72) Ala-Ser-Pro-Asp-Tyr-Arg yi = S gyöksúly + 19 A fragmentáció módszer- és készülék-függő!
-CO-NH-CH(R2)-CO~ ~CO-NH-CH(Rn)-COOH. yn-2. bn-1. yn-1. y1. b2. bi=S gyöksúly + 1. (72) Ala-Ser-Pro-Asp-Tyr-Arg yi = S gyöksúly A fragmentáció módszer- és készülék-függő!")
52
SHREK - Elméleti MALDI-CID spektrum – MS product
60.04 S 130.09 y1-NH3 276.16 y2 399.21 b3+H2O 528.25 b4+H2O 70.07 R 138.07 H 277.14 HR-NH3 406.18 HRE-NH3 552.29 y4-NH3 84.08 K 147.11 y1 286.15 RE 415.23 y3-NH3 556.26 m3 87.09 197.10 a2 294.17 HR 423.21 HRE 569.32 y4 100.09 207.09 b2-H2O 336.18 a3-NH3 432.26 y3 575.30 m2 101.11 225.10 b2 353.20 a3 465.22 a4-NH3 583.32 m4 102.06 E 258.16 RE-28 363.19 b3-H2O 482.25 a4 584.27 m5 110.07 259.13 y2-NH3 364.17 b3-NH3 492.23 b4-H2O 625.33 m1 112.09 266.17 HR-28 381.20 b3 493.22 b4-NH3 129.10 269.12 RE-NH3 395.22 HRE-28 510.24 b4

53
SHREK – elméleti ECD fragmensek
z1 c2 a3 z3 c4 a2 z2 c3 a4 z4 Elméleti „spektrum”: → lehetséges fragmensek, egyenlő intenzitással

54
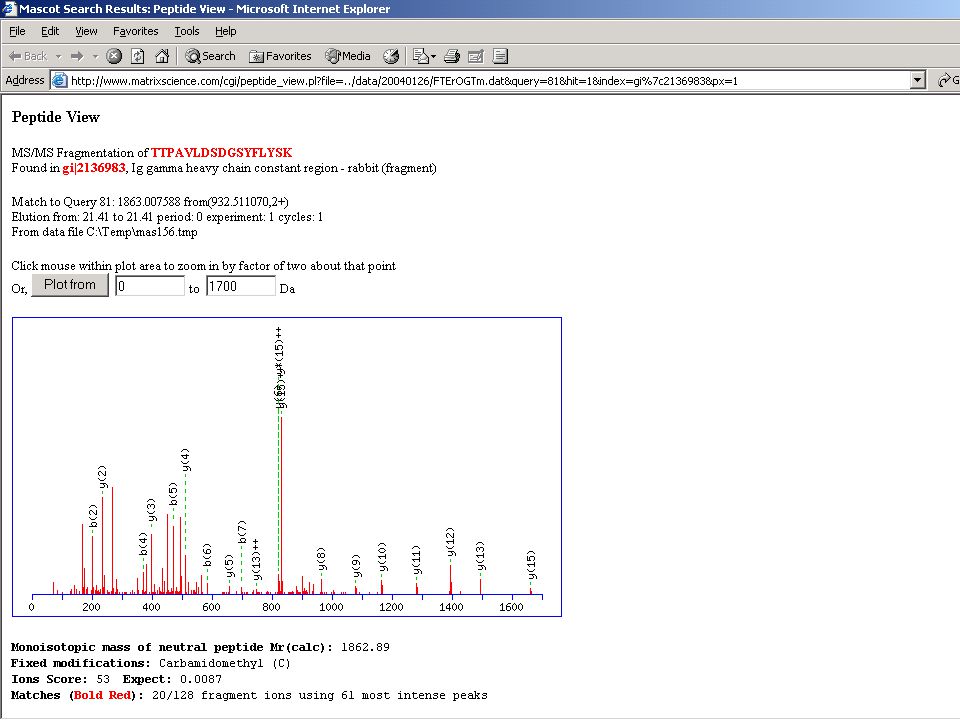
És egy valóságos spektrum

55
A proteomika „Achilles sarka”?
Elméleti azaz megjósolt „adatokon” alapuló azonosítás

56
bioinformatika = bio + informatika
Nem szabad elfelejtenünk, hogy biológiai mintákkal dolgozunk! és kémiailag is aktív vegyületekkel! Az algoritmus lehet tökéletes, csak a biológiai rendszer nem viselkedik!

57
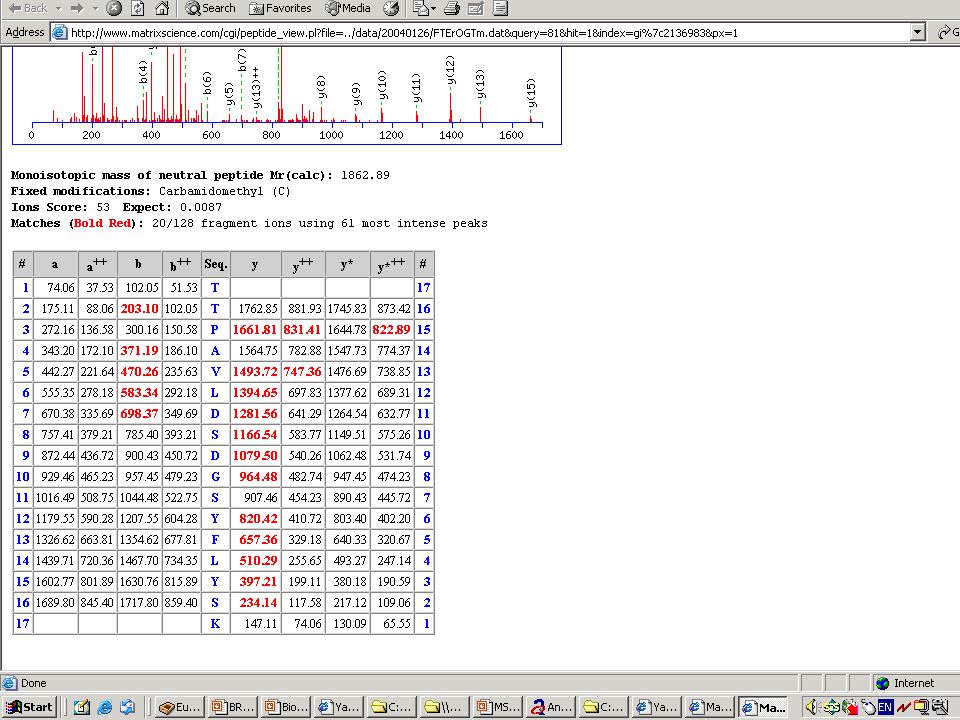
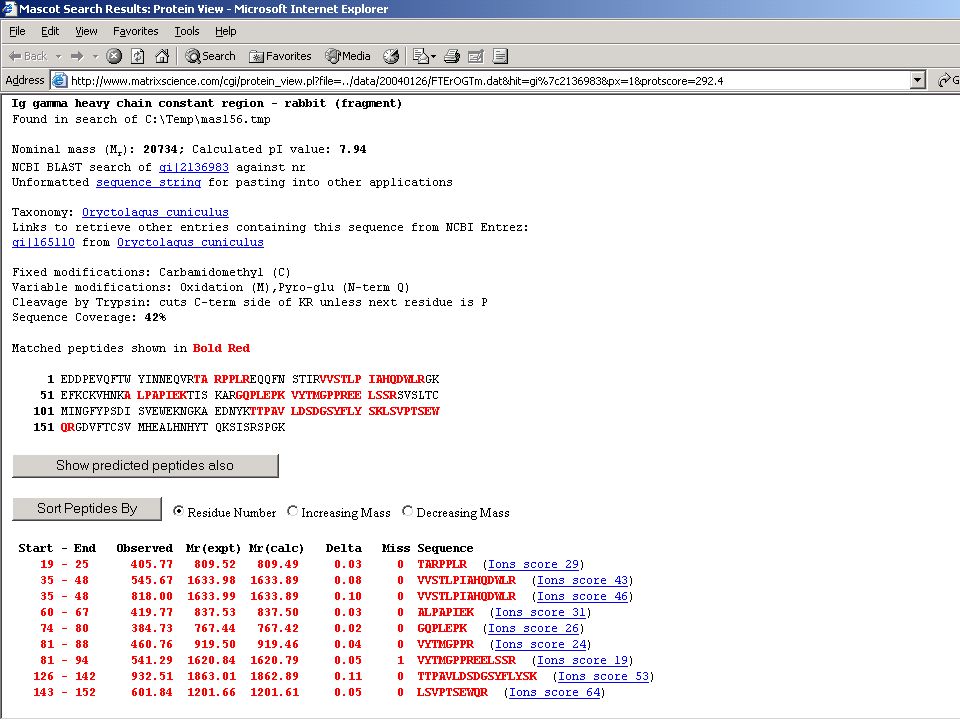
Megtörtént a frakcionálás, az emésztés...Jön az analízis!
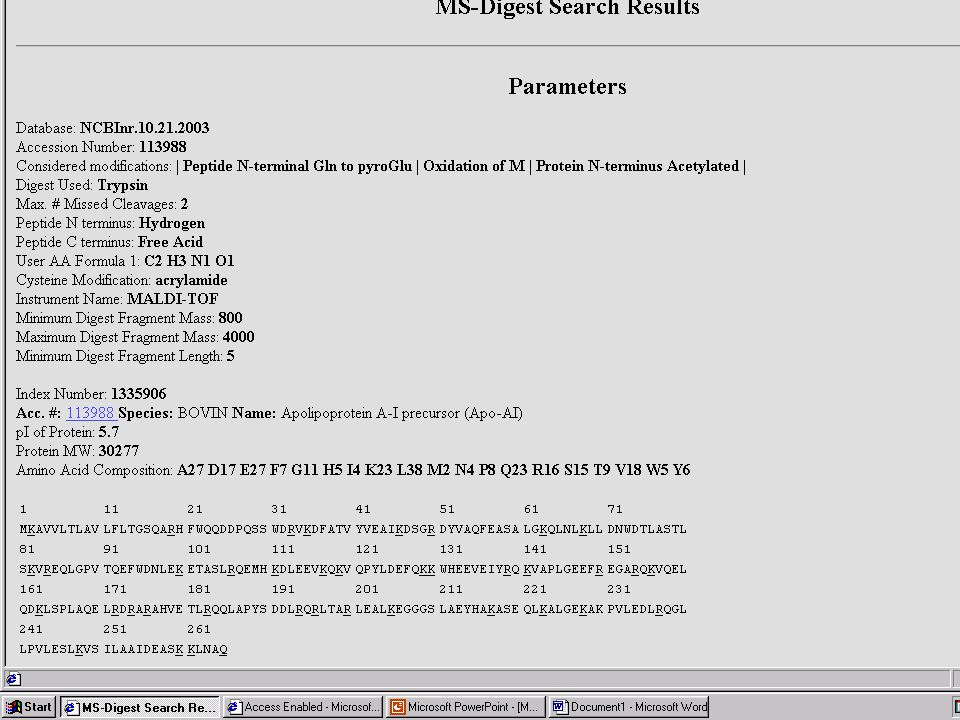
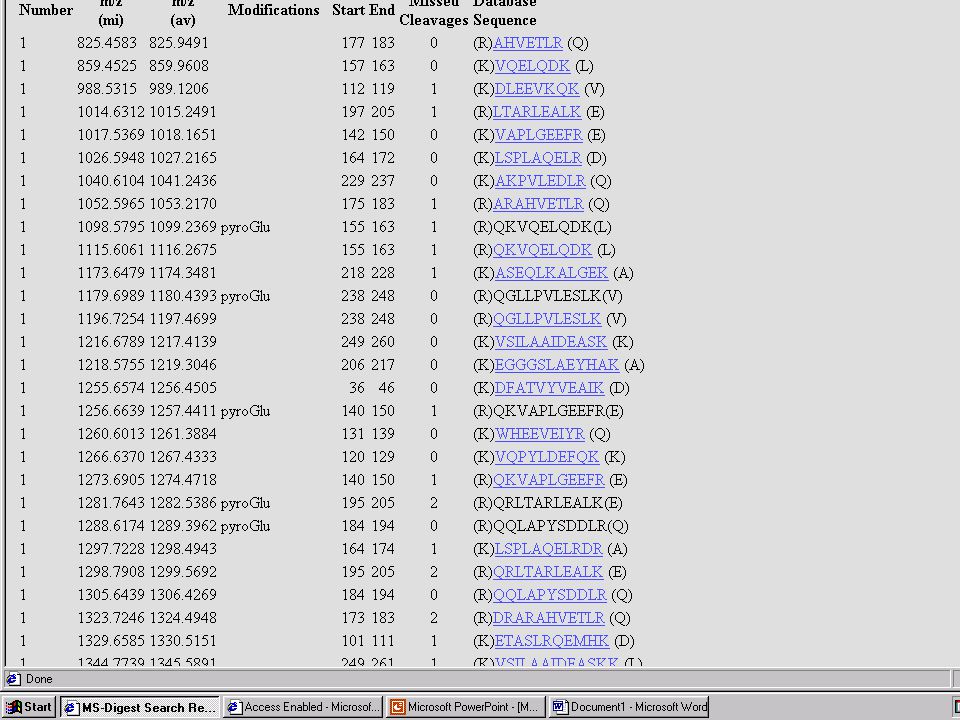
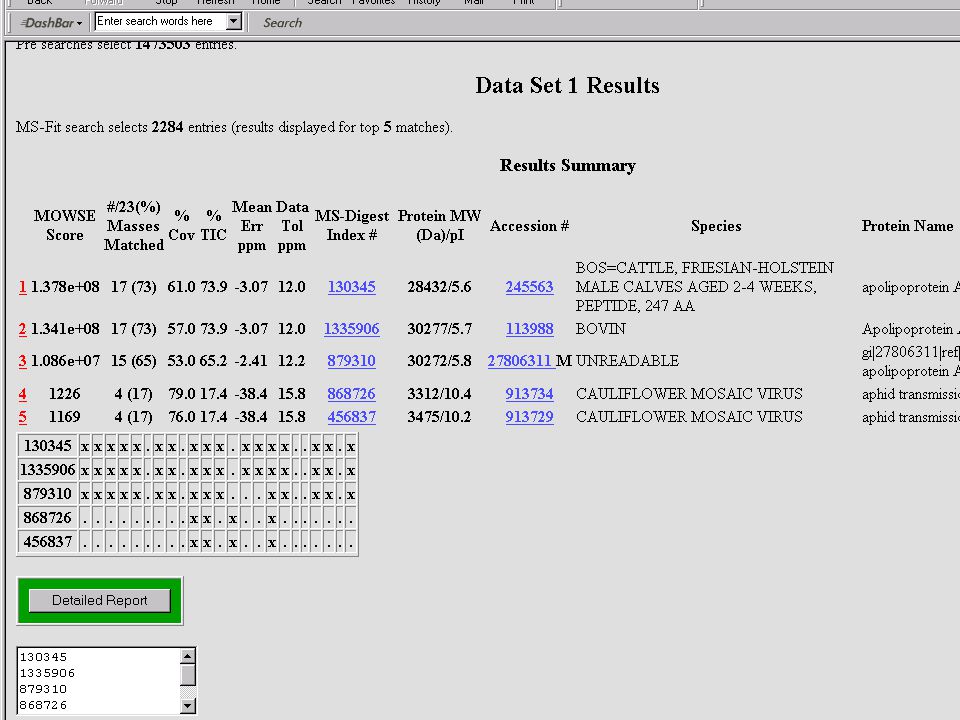
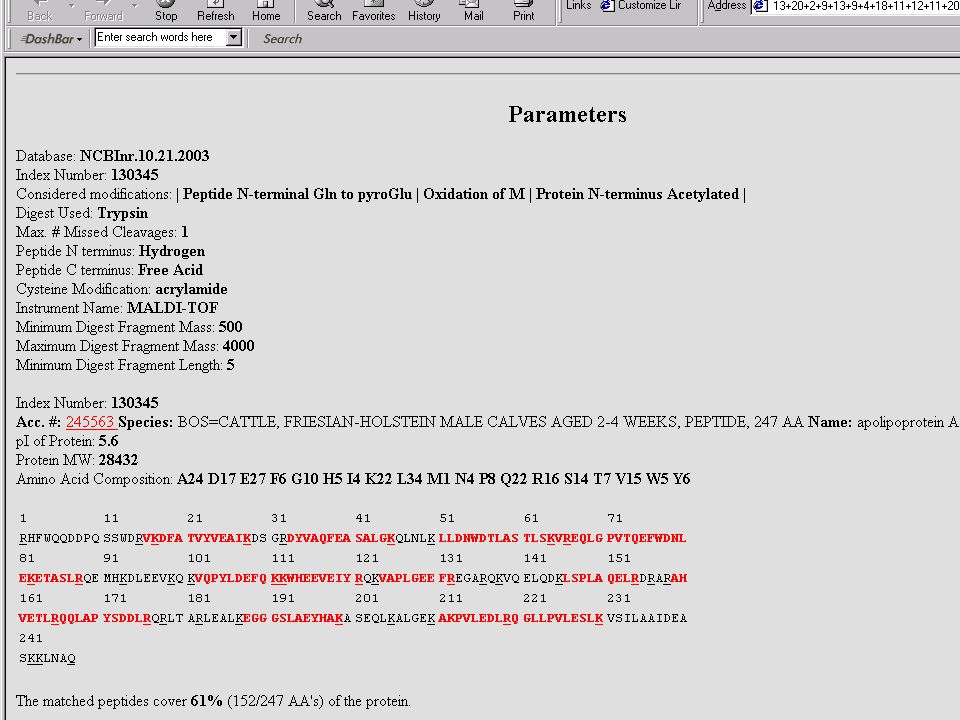
Mondjuk, MALDI MS-mérés frakcionálatlan, vagy HPLC után → egy jellemző (?) MH+ sorozat ↓ MS-Fit Peptide Mass Fingerprint – alapú fehérjeazonosítás Mi lehet a baj? → aspecifikus hasítás, kovalens módosítás, szennyezés stb.
 MH+ sorozat. ↓ MS-Fit. Peptide Mass Fingerprint – alapú fehérjeazonosítás. Mi lehet a baj → aspecifikus hasítás, kovalens módosítás, szennyezés stb.")
63
Nem mindig ilyen szép a menyasszony!
azaz a PMF-alapú azonosítást meg kell erősíteni MIÉRT??? – lásd előbb és később is... a „vád tanúja”: az MS-MS kísérlet eredménye

64
MS-MS eredményekkel is lehet keresgélni!

65
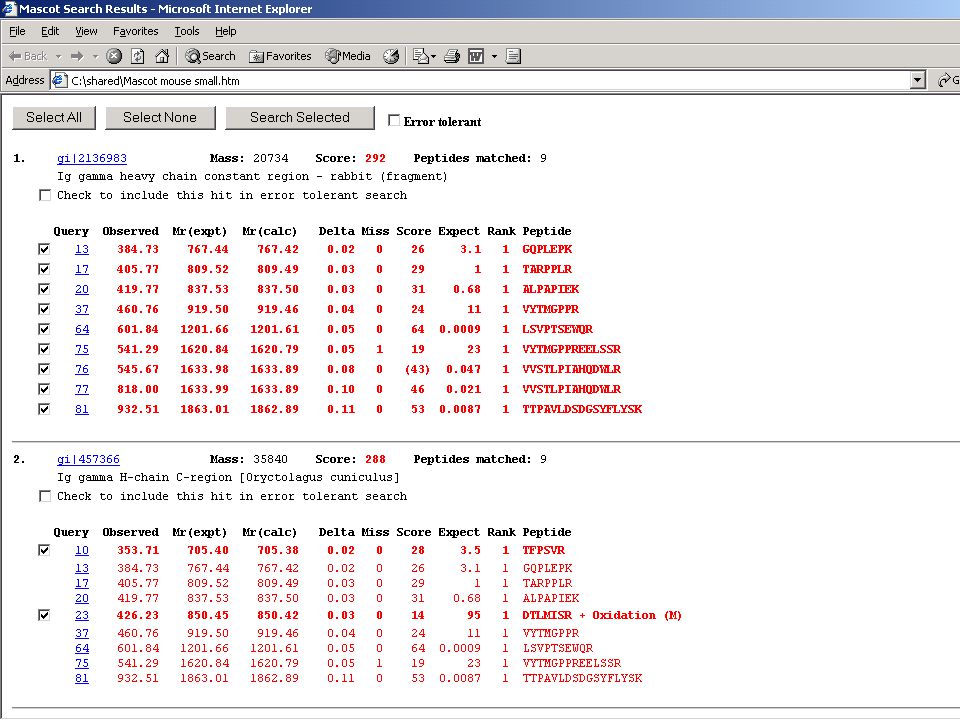
Független PMF és MS-MS azonosítás az igazi!

66
Elegánsabb az on-line LCMSMS
A nanoHPLC oszlopról minden megy a tömegspektrométerbe (250 nL/min) ún. „adatfüggő” adatgyűjtés MS mérés Komputer kiválasztja a legintenzívebb iont MS-MS mérés
 ún. „adatfüggő adatgyűjtés. MS mérés. Komputer kiválasztja a legintenzívebb iont. MS-MS mérés.")
67
MS + MS-MS combined total ion current

68
Lekeresés csak MS-MS adatokkal
„script” – az összes MS-MS adatok megfelelő formába rendezve az egyes spektrumokkal független lekeresés az eredményeket viszont fehérjénként összegezzük

71
Nem igazi adatokhoz, hanem az elméleti spektrumhoz hasonlítunk!

75
Kovalens módosítások detektálása
szerencsés esetben találhatunk ilyet is automatizált MS-MS analízissel lekereső programok lehetővé teszik a kovalens módosítások követését

76
Túl sok variációs lehetőség
rengeteg félreértelmezést okozhat !

77
Schistosoma mansoni proteomikája G. Knudsen, J.H McKerrow, UCSF
* kb. 200 millió ember fertőződik évente, bőrön keresztül, fertőzött vízben * Átmeneti hordozó – Biomphalaria glabrata * Kulcs a fertőzéshez: a cercaria által kiválasztott enzim-keverék

78
Egyszerű, jól ismert keverékkel van dolgunk...
a csigából kibújt féreg-növendékeket jól lemossuk bőr-lipidekkel megindítjuk a kiválasztást a fehérje-oldatot bekoncentráljuk 1D SDS-PAGE fehérje-frakcionálásra gélben emésztés tripszinnel LC/MS/MS analízis Eksigent/Famous - 75 mm ID PepMap column; 300 nL/min flow; water/ACN/0.1% HCOOH mobile phase, gradient elution, QSTAR; 1 sec MS/3 sec CID; single charged and dynamic exclusion

79
Mit találtunk? cercaria felszíni fehérjéket
cercaria elasztázt és glikolitikus enzimeket csiga immunválasz-fehérjéket fotoszintézisben résztvevő fehérjéket humán keratint marha szérum albumint

80
Honnan jött mindez? a cercaria külsejéről ez az igazi! – a kiválasztott fehérjék az átmeneti hordozóból a salátából, amit a csigák ettek – éheztessük őket? a technikus kezéről? hajából? a laborból mindenütt – lehet, hogy takarítani kellene? Új, hatásosabb izolálási módszer→ az elasztáz-aktivitás 40x-esére nőtt!

81
Eddig csak kvali volt... Kvantitatív proteomika
1) a frakcionálás szintjén Pl. 2D gélek összehasonlítása vizuálisan, komputer programokkal, differenciál festéssel 2) az MS-analízis során
 a frakcionálás szintjén. Pl. 2D gélek összehasonlítása vizuálisan, komputer programokkal, differenciál festéssel. 2) az MS-analízis során.")
82
Az MS NEM kvantitatív módszer
* a különböző molekulák nem egyformán detektálhatók pl. különböző peptidek relatív intenzitása nem tükrözi relatív mennyiségüket * nem minden komponenst detektálunk az elegyekből

83
Hogy lehet ezt legyőzni?
csak nagyon hasonló peptidek relatív intenzitását vethetjük össze! 1) Jelöljünk két sejt-populációt hasonlóan, de megkülönböztethetően! 2) Keverjük össze az összes fehérjét! 3) A keveréket emésszük, analizáljuk! És így kvantizhatunk!
 Jelöljünk két sejt-populációt hasonlóan, de megkülönböztethetően! 2) Keverjük össze az összes fehérjét! 3) A keveréket emésszük, analizáljuk! És így kvantizhatunk!")
84
Hogyan jelölhetünk? ICAT – Cys-oldalláncának alkilezése, könnyű (H8) és nehéz (D8) alkil-csoporttal → amiben nincs Cys, az kiesik → a deuterált vegyület retenciós ideje kicsit hosszabb azonosítás: MS-MS alapon kvanti: MS-alapon relatív intenzitásokból ingadozás: van az 30% is!

85
Tripszines emésztéshez Arg a menő!
Hogyan jelölhetünk? SILAC (stable isotope labeling) C13, N15, O18 tápanyaggal visszük be – jelölt aminosav → Olyat kell választani, ami nem alakulhat át más aminosavvá, illetve nem szintetizálódik a sejtben Tripszines emésztéshez Arg a menő! a keveréket frakcionáljuk, emésztjük, analizáljuk azonosítás MS-MS alapon kvanti a relatív intenzitásból
 C13, N15, O18. tápanyaggal visszük be – jelölt aminosav. → Olyat kell választani, ami nem alakulhat át más aminosavvá, illetve nem szintetizálódik a sejtben. Tripszines emésztéshez Arg a menő! a keveréket frakcionáljuk, emésztjük, analizáljuk. azonosítás MS-MS alapon. kvanti a relatív intenzitásból.")
86
Hogyan jelölhetünk? tripszines emésztés H2O18-ban
Két oxigén lép be, minden peptid jelölődik Rengeteg információ Még a szekvenálás is könnyebb, a C-terminális ionok csúsznak DE beszárítás a normál emésztés után, hosszú enzimes inkubáció... → veszteségek, nem specifikus hasítások

87
Hogyan jelölhetünk? iTRAQ ™ (Applied Biosystems)
Nagyon ravasz jelölés! amino-csoportra (N-terminus, Lys) 4-féle stabil izotópos szerkezet, azonos additív tömeggel, de különböző fragmensekkel (m/z 114, 115, 116, 117) azonosítás és kvanti az MS-MS adatokból!
 4-féle stabil izotópos szerkezet, azonos additív tömeggel, de különböző fragmensekkel (m/z 114, 115, 116, 117) azonosítás és kvanti az MS-MS adatokból!")
88
iTRAQ jelölés Ross et. al. MCP 2004
4 mintát kvantitatíve megkülönböztetni. Minden peptidet jelöl, a módosítottakat is Egyszerű a mintaelőkészítés Jelnövelő hatású (szuperponálódik a 4-féle jel) 20%-nál kisebb standard deviáció Jobb minőségű CID spektrumok Poszt-transzlációs változásokat is lehet így mérni! DRÁGA Ross et. al. MCP 2004
 20%-nál kisebb standard deviáció. Jobb minőségű CID spektrumok. Poszt-transzlációs változásokat is lehet így mérni! DRÁGA. Ross et. al. MCP")
89
Mi ebből a tanulság?/ Take home message
Ha biológus vagy → eredj programozást és statisztikát tanulni Ha matematikus vagy → irány a biológia Ha proteomikával akarsz foglalkozni → tömegspektrometria sem árt

90
Mi ebből a tanulság?/ Take home message
Mennyiben lehet biológiai folyamatokat statisztikailag/matematikailag elírni? A komputer gyors, nem hibázik, „tanulékony”, de hülye... Mi emberek ellenben képesek vagyunk felismerni a váratlant, az újat – feltéve, hogy használjuk az eszünket....

91
Minta kérdések Mitől függ, mennyire jó egy adatbázis?
Mi az oka, hogy nincs tökéletesen működő adatbázis-lekereső szoftver? Miért van szükség proteomikára? Miért kell „jelölni” kvantitatív proteomikában? Miért előnyös egy olyan jelölés, ami minden peptidet módosít? És valóban módosít-e minden peptidet?

92
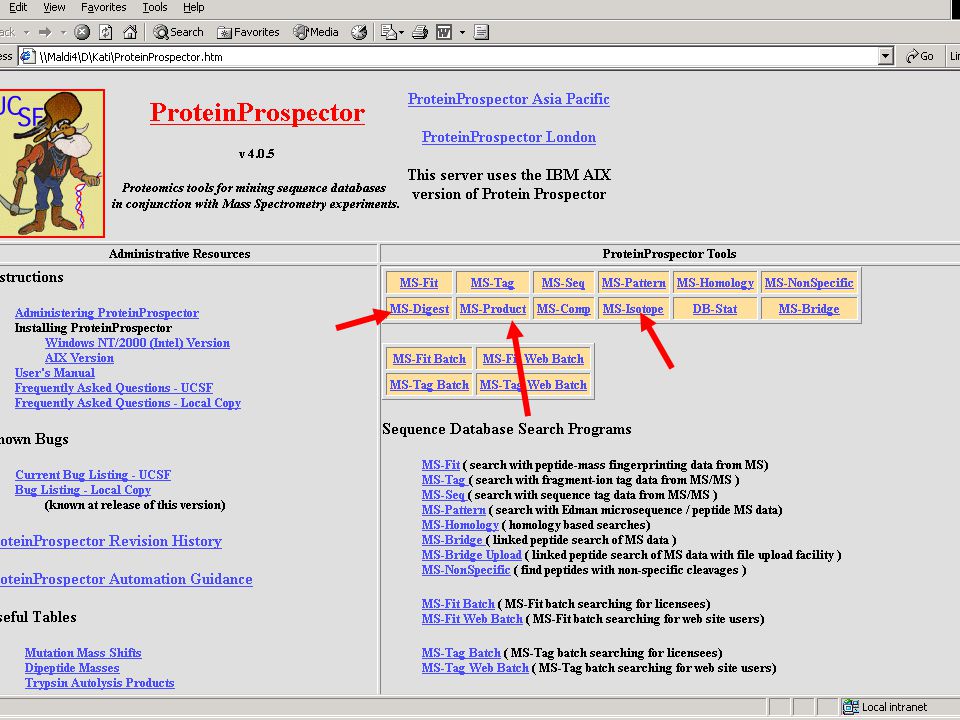
Hasznos web-oldalak ProteinProspector – http://prospector.ucsf.edu
MS-BLAST - BLAST - European Bioinformatics Institute - Szekvencia-összehasonlítás – MS kezdőknek - „What is mass spectrometry” -

Hasonló előadás











>")





 7) 8) 9) 10) Mennyi az x, y és z értéke? 11) 12) 13) 14) 15)>")





