Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
MOLEKULÁRIS GENETIKAI MUTATIOS ANALYSIS ENZYMOPATHIÁKBAN (Gaucher kór, Fabry-, Homocystinuria. Menkes-kór, Wiscott–Aldrich syndroma) László Aranka, Amparo Chabas, Endreffy Emőke, Várkonyi Ágnes, Timár Erzsébet, Sztriha László, Viktor Kozich, H.K. Ploos van. Amstel, Zeynep Tümer, Hans Ochs, Brian Smart, Raskó István SZTE Szent-Györgyi Albert Orvostudományi Egyetem, Gyermekklinika, Inst. Biochim. Clinica, Barcelona, Charles Univ. First Faculty of Medicine, Center for lnherited Metab Praha, Czech Republic, DNA Laboratory KGCU Univ. Hosp. Utrecht. Netherlands, J.F. Kennedy Institute, Glostrup, Denmak, Division of Infectious Disease, Immunology and Rheumathology, Washington, MTA, SZBK, Genetikai Intézet, Szeged
 László Aranka, Amparo Chabas, Endreffy Emőke, Várkonyi Ágnes, Timár Erzsébet, Sztriha László, Viktor Kozich, H.K. Ploos van. Amstel, Zeynep Tümer, Hans Ochs, Brian Smart, Raskó István SZTE Szent-Györgyi Albert Orvostudományi Egyetem, Gyermekklinika, Inst. Biochim. Clinica, Barcelona, Charles Univ. First Faculty of Medicine, Center for lnherited Metab Praha, Czech Republic, DNA Laboratory KGCU Univ. Hosp. Utrecht. Netherlands, J.F. Kennedy Institute, Glostrup, Denmak, Division of Infectious Disease, Immunology and Rheumathology, Washington, MTA, SZBK, Genetikai Intézet, Szeged.")
2
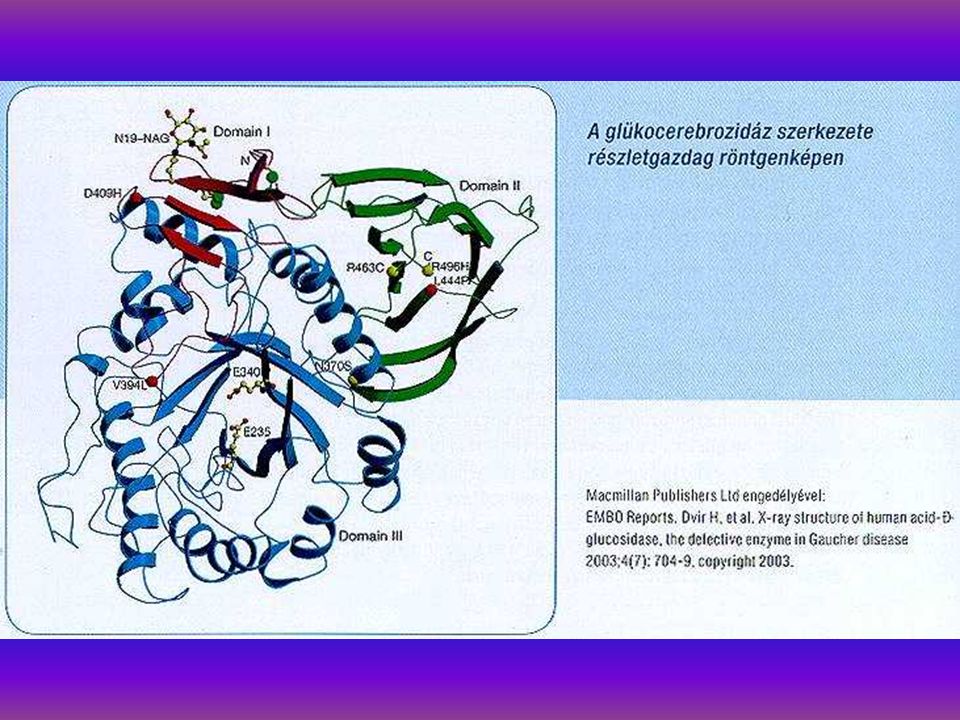
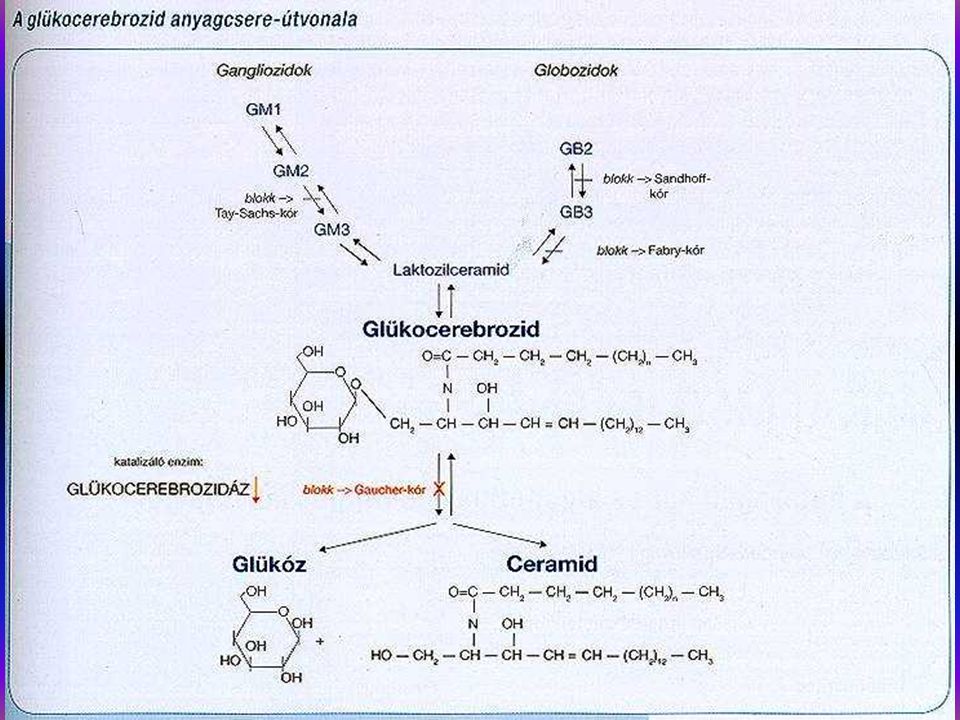
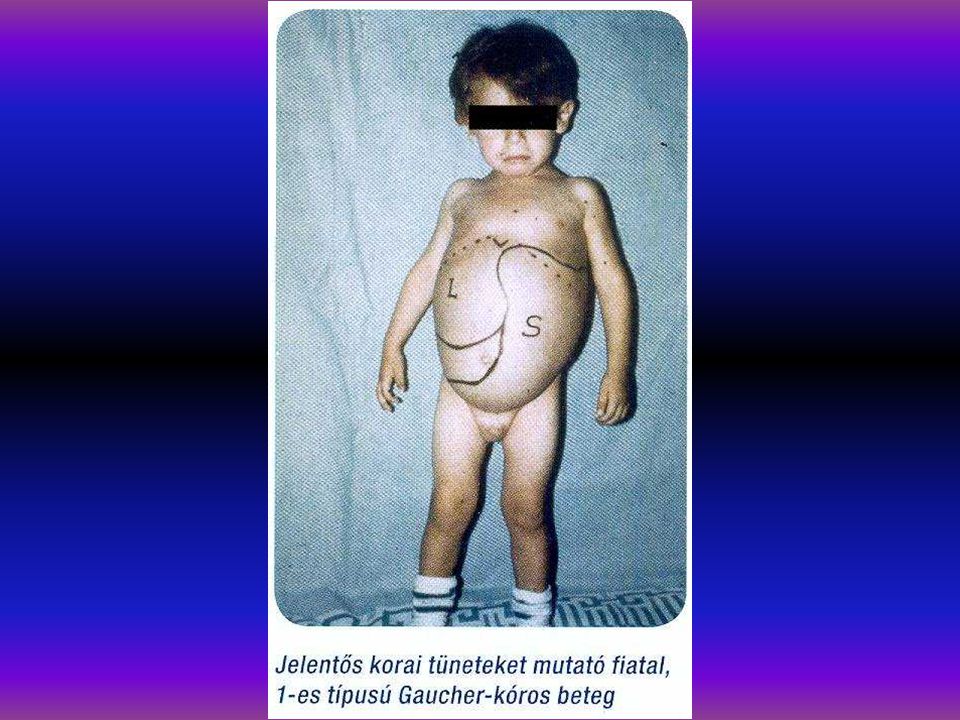
A Gaucher kór autosomalis recessiv örökletes kórkép, glycolipid raktárbetegség, glucocerebrosidase hiány következtében (glucocerebrosidase gén 1q21). 15 hazai Gaucher kóros családban végeztünk leukocyta- homogenisatumból specifikus enzmdiagnózist, glucosidase aktivitás meghatározást a homo- és heterozygotaság kiszűrésére. 15 homozygotát és 11 G. heterozygotát diagnosztizáltunk. DNS izolálásra és molekuláris genetikai diagnosztikára 15 családban került sor. N370S/de155. N370SIL444P, N370S/7 genotypusokat detectáltunk. Irodalmi adatok szerint az N370S mutatio a non neuropathiás forma, spanyol populatioban 44,3%. 625 É- Portugál, 38,9% UK-ban. Az L444P mutatio ugyancsak neuropathiás Gaucher-ban található (Tsuji et al 1987). Enyhe mutatiok N370S, R359Q, R496H.
. Enyhe mutatiok N370S, R359Q, R496H..")
8
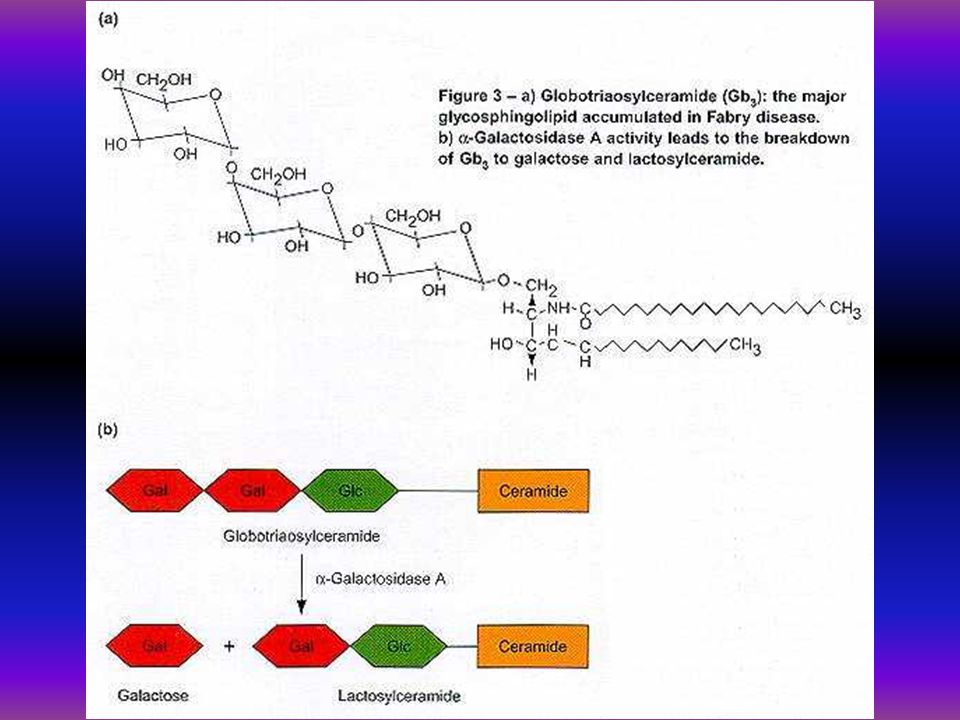
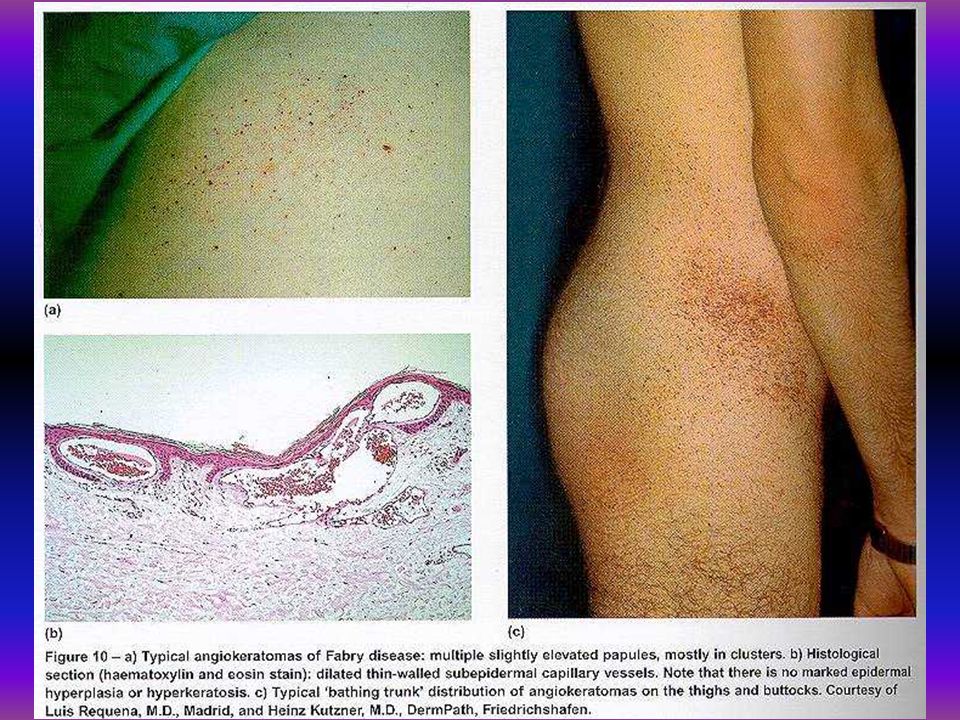
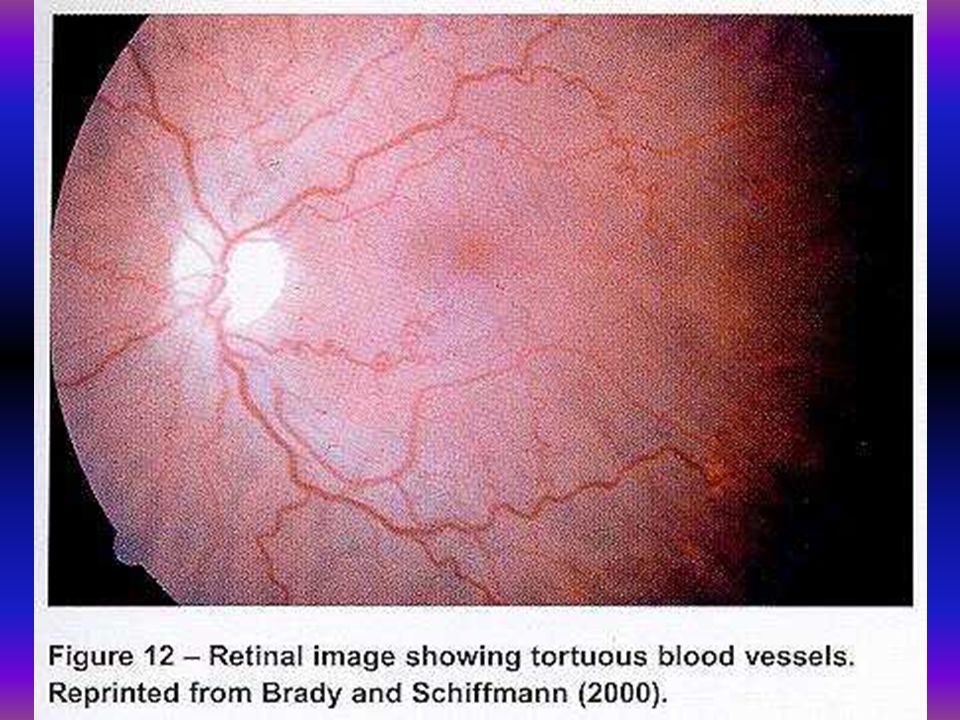
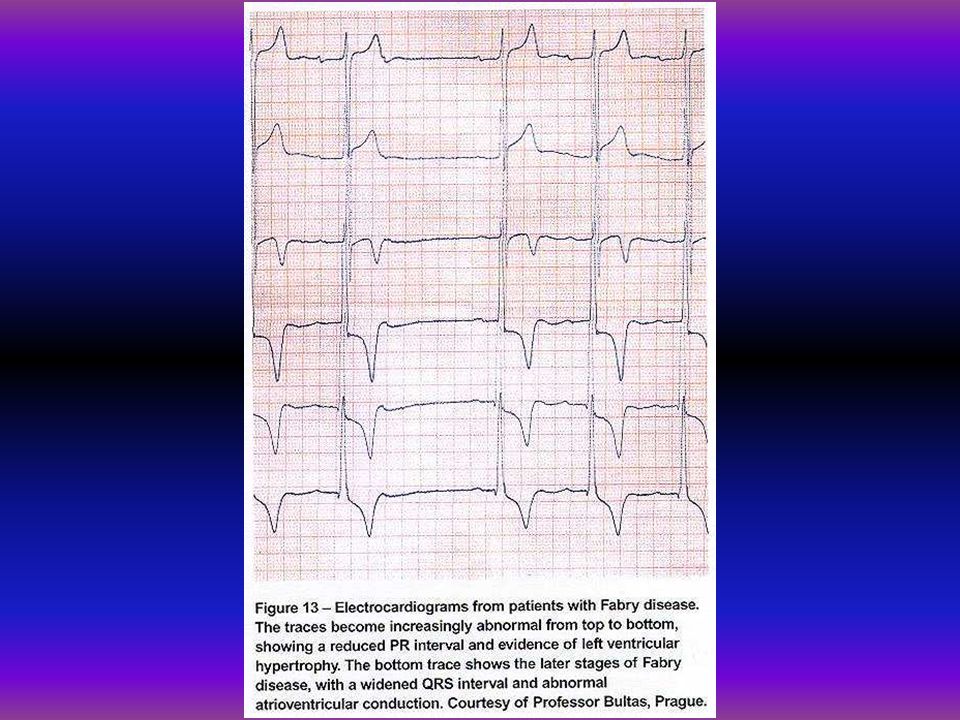
Mutatios analysis Fabry-kórban 19 éves férfibetegünknél biokémiai és molekuláris vizsgálat történt. Az alpha-galactosidase aktivitás 1,86 nmol/mg/h volt, az anya és a beteg húga génhordozó 36 nmol/mg/h, 21,7 nmol/mg/h, kontroll 130 nmol/mg/h. Az alpha-galactosidase gén 5. exonjában missense mutatio igazolódott a 266-os codonban az asparaginsavat tyrozin pótolta G-T transversio a 10287 positioban.

16
Homocystinuria Cystationine beta synthase (CBS) gén alábbi 4 mutatióját vizsgáltuk PCR/RFLP technikával (Sebastio et al Hum. Genet. 56: 1324-1333, 1995). 16 éves fiúbetegünknél a plasma és vizelet homocystin szintje extrémfokban emelkedett. Mutatios eredmények: P145L -/- R125Q: -/- L278T: +1-G307S: -/-, compound heterozygotának bizonyult.
. 16 éves fiúbetegünknél a plasma és vizelet homocystin szintje extrémfokban emelkedett. Mutatios eredmények: P145L -/- R125Q: -/- L278T: +1-G307S: -/-, compound heterozygotának bizonyult..")
17
Homocystinuria

18
Menkes gén mutatiós analysise (ATP7A) Módszer: Dideoxy finger printing DDFP és PCR 12-es exon direct sequenálása. Eredmény: Bázispár substitutio missense mutatioval. Wiscott–Aldrich sy. molekuláris genetikai diagnosztikája, praenatalis diagnózisa A Wiscott–Aldrich gén (WAS) az Xp11.22-p 11.23-ra localizálódik. 1820 bázispárból áll. A mutatios analysis mRNA/cDNA felhasználásával történik. L. L. 8 éves fiúbetegünk esetében két bázispár deletio igazolódott (313G, 314T) frame shift-et eredményezve és STOP-ot a 120-as (ASP) codonban. Az anya és az anya húga ugyanezen mutatiora carriernek bizonyult. 1 normál allélt és két bázispár deletioból egy allélt hordoznak. Praenatalis dg.: Chorionboholyból a fiú foetus esetében kizárható volt a fenti molekuláris módszerrel a hemizygota érintettség. Conclusio: Fenti kórképekben az alkalmazott molekuláris biológiai metodikák biztonságos diagnózist adnak, praenatalis kórismére is alkalmasak a fenti, jórészt gyógyíthatatlan kórképekben.
 az Xp11.22-p ra localizálódik bázispárból áll. A mutatios analysis mRNA/cDNA felhasználásával történik. L. L. 8 éves fiúbetegünk esetében két bázispár deletio igazolódott (313G, 314T) frame shift-et eredményezve és STOP-ot a 120-as (ASP) codonban. Az anya és az anya húga ugyanezen mutatiora carriernek bizonyult. 1 normál allélt és két bázispár deletioból egy allélt hordoznak. Praenatalis dg.: Chorionboholyból a fiú foetus esetében kizárható volt a fenti molekuláris módszerrel a hemizygota érintettség. Conclusio: Fenti kórképekben az alkalmazott molekuláris biológiai metodikák biztonságos diagnózist adnak, praenatalis kórismére is alkalmasak a fenti, jórészt gyógyíthatatlan kórképekben..")
19
Molekularis genetikai mutatios analysis Canavan- és Hunter kórban László A., Jakobs C., Shaag A., Elpeleg O.N., Horváth K., Gál A., Suzanna Bunge, Endreffy E., Szabó M., Raskó I. Szegedi Tudományegyetem ÁOK, Gyermekklinika, Gyermekklinikai Laboratórium, Free University Hospital, Amsterdam, Shaare–Zedek Medical Centre Metabolit Unit. Jerusalem, ÁOK, Radiológiai Klinika, Szeged, SZBK, Genetikai Intézet, Szeged

20
A Canavan kór (CD) progresszív, infantilis typusú, autosomalis recessiven öröklődő neurodegenerativ kórkép leukodystrophia, amely rapid súlyos lefolyású, motoros functiokésés az érdeklődés elvesztése, spasticitás megalocephalia, progressiv vakság. Pathológiailag myelinvesztés, az agyszövet korai halálhoz vezető szivacsos degeneratioja. Biokémiailag N-acetyl-L-aspartat-amido-hydrolase (ASPA) enzymdefektus jellemzi, amely a központi idegrendszerben asparaginhiányt indukál. Beteg: 7 hónapos csecsemő, progressiv cerebralis laesioval, jellegzetes craniális. MRI képpel. Ideg és izombiopsia nem volt kórjelző.
 enzymdefektus jellemzi, amely a központi idegrendszerben asparaginhiányt indukál. Beteg: 7 hónapos csecsemő, progressiv cerebralis laesioval, jellegzetes craniális. MRI képpel. Ideg és izombiopsia nem volt kórjelző..")
21
Módszer: Specifikus enzymvizsgálat a normális arylsulphatase- A activitás (45,8 nmol/mg/protei/h) heterozygotaság alapján metachromatikus leukodystrophia homozygota érintettséget kizárt, a norm. galactocerebrosid- beta-galactosidase activitás orthochromatikus leukodystrophiát vetett el. Kórosan emelkedett a vizelet N- acetylaspartat sav (165 nmol/mol/kreatinin [norm.: 6,6–34]), mely Canavan kórt bizonyított.
, mely Canavan kórt bizonyított..")
22
Molekuláris genetikai vizsgálatok: A genomikus DNS-t peripheriás vérből standard módszerrel extraháltuk. A Canavan gén 6-os exonjának 183 bp fragmensét amplifikáltuk a genomikus DNS-ből, PCR 2 oligonucleotid primert használtunk CDP1 (CTCTTGATGGGAAGACGATC és CDP2) (ACACCGTGTAAGATGTAAGC). A PCR produktumot direkt sequenáltuk fluorescens dideoxy lánc terminális reakcióval és ABI 377 automata sequenálóval. Eredmények: 914 (A305 E) mutatiora homozygotaság igazolódott a csecsemőnél, ezen mutatiora. A heterozygota frequentia 1/35 és 1/59 az Askenasi populatioban (Kupietzky és mtsai 1988). Ezen mutatio gyakori Európa-szerte a nem-Askenasi populatiokban, a mutált alléloknak kb. 40%-a.
 (ACACCGTGTAAGATGTAAGC). A PCR produktumot direkt sequenáltuk fluorescens dideoxy lánc terminális reakcióval és ABI 377 automata sequenálóval. Eredmények: 914 (A305 E) mutatiora homozygotaság igazolódott a csecsemőnél, ezen mutatiora. A heterozygota frequentia 1/35 és 1/59 az Askenasi populatioban (Kupietzky és mtsai 1988). Ezen mutatio gyakori Európa-szerte a nem-Askenasi populatiokban, a mutált alléloknak kb. 40%-a..")
23
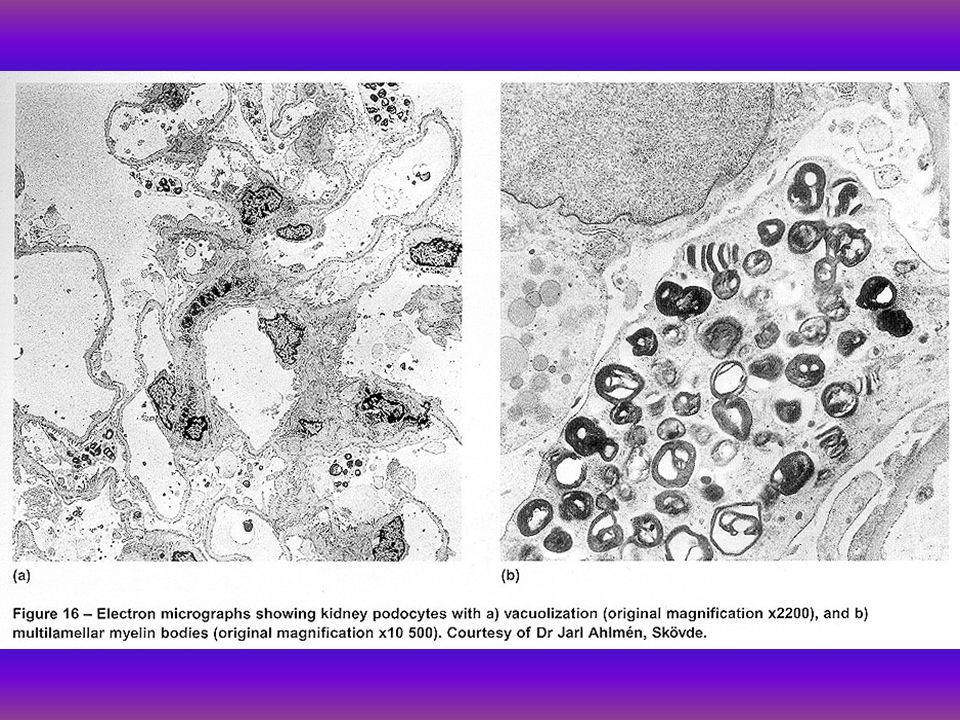
Molecularis genetikai mutatios analysis Hunter kórban A Hunter kór iduronat-2-sulphatase (IDS EC3.1.6.13) X-hez kötött recissiv örökletességű lysosomalis kórkép, heparan és dermatan tárolással. A betegek garoyloid phenotypusuak skeletalis dysmorrphia, ízületi merevség, hepatomegalia, cardiovascularis elégtelenség és mentalis retardatio dominál. Az IDS gén 23 kB 550 aminósavat kódol (Wilson et al 1990.), a genomikus structurát (Flomen és mtsai 1993, Wilson 19934, Lu 1994) közölte 4 hazai Hunter syndromás beteg molekuláris genetikai mutációs analysisére került sor. Módszer: Elsőként Southern-Blot kivitelezése történt. cDNA próbával és a betegek genomikus DNS-ét Hind III. Stu L, TaqI restrikciós endonucleázokkal emésztettük. PCR/RFLP történt, ha az RFLP nem volt informativ, SSCP analysis és direct sequenálásra került sor (Bunge és mtsai 1992). Eredmények: MPS II betegekben kórosan fokozott glucosaminuriát találtunk dysostosissal, jellemzőek voltak a szövettani, fény- és elektronmikroszkópos leletek conjuctiva biopsiából. Missence, non-sencc. splice és frame- shift mutatiokat detektáltunk.
, a genomikus structurát (Flomen és mtsai 1993, Wilson 19934, Lu 1994) közölte 4 hazai Hunter syndromás beteg molekuláris genetikai mutációs analysisére került sor. Módszer: Elsőként Southern-Blot kivitelezése történt. cDNA próbával és a betegek genomikus DNS-ét Hind III. Stu L, TaqI restrikciós endonucleázokkal emésztettük. PCR/RFLP történt, ha az RFLP nem volt informativ, SSCP analysis és direct sequenálásra került sor (Bunge és mtsai 1992). Eredmények: MPS II betegekben kórosan fokozott glucosaminuriát találtunk dysostosissal, jellemzőek voltak a szövettani, fény- és elektronmikroszkópos leletek conjuctiva biopsiából. Missence, non-sencc. splice és frame- shift mutatiokat detektáltunk..")
24
Mutation analysis of the Menkes disease gene (ATP7A) Menkes disease (MD) is an X-linked recessive lethal disorder of copper metabolism with progressive neurodegeneration and connective tissue disturbances leading to disfunction of some copper enzymes (COX) Danks 1995. MD locus, ATP7A is mapped to Xq 13.3 (Verga et al,1991, Tümer 1992) ATP7A is organised into 23 exons spanning a genomic region of 150 kb; the exons are relatively large, 77 bp- 726 bp of coding sequence
 ATP7A is organised into 23 exons spanning a genomic region of 150 kb; the exons are relatively large, 77 bp- 726 bp of coding sequence.")
25
Mutations leading to MD show great variety, cytogenetic abnormalities, partial deletions of ATP7A intrachromosomal rearrangement balanced X-autosomal translocation,X;2, X;21 In 20% of 200 patients screened there were found gross deletions or rearrangements (Southern-blott) or by PCR amplification of exons/intron- specific primers (Z. Tümer, N. Horn,1996) 12 point mutation have been reported with severe phenotype (Das et al,1994, Tümer et al, 1996)
 12 point mutation have been reported with severe phenotype (Das et al,1994, Tümer et al, 1996).")
26
Galactosaemia Dr. László Aranka SZTE Gyermekklinika

27
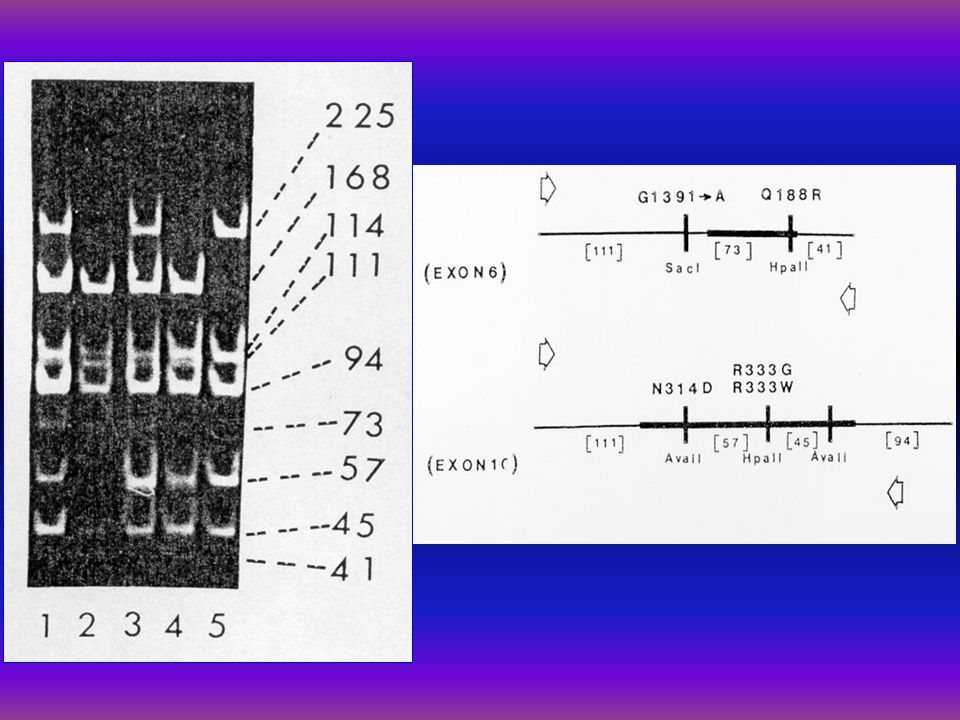
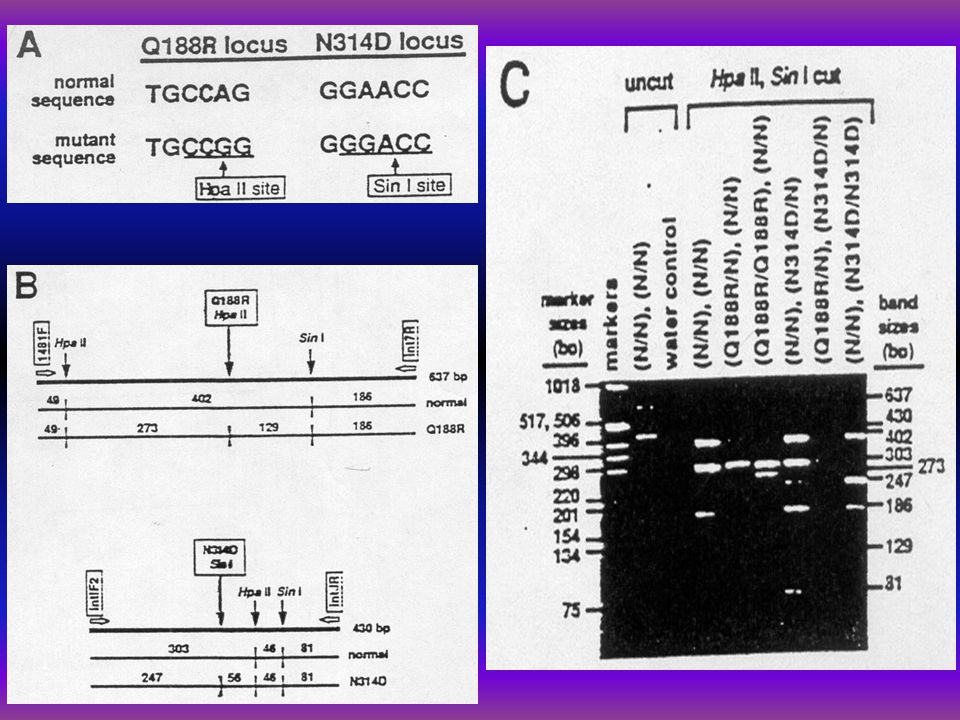
Genotypes in Hungarian galactosemic patients A. László l, Z. Havass 2, E. Endreffy l, P. Megyeri 1, L. J. Elsas 3, Yoon Shin 4, T. Podskarbi 4 l Dept. of Pediatrics, A. Szent-Györgyi Med. Univ., Szeged, 2 Dept. of Pediatrics Erzsébet Hospital, Hmvhely, Hungary, 3 Dept. of Pediatrics, Emory Univ. School of Med. Atlanta, USA, 4 Dept. of Pediatrics and Med. Immunol. Lab. Dr. von Hauner Hosp., Munich, Germany l. Background: In the main galactosemic mutation (Q188R) the arginine is substituted for glutamine at codon 188 in exon 6, with nearly 70% prevalence in Caucasians with galactosemia. In the Duarte mutant allele of the galactose-1-phosphate uridyl- transferase GALT, EC 2.7.7.12.) GALT genome in base pair 2744 of exon 10 there is an A to G transition resulting an acidic aspartate (D) substitution for an asparagine (N) at codon 314 (N314D) (Reichardt et al. 1992). 2. Subjects: Genotypes of GALT deficient Hungarian galactosemic patients were determined as a population genetic study. 3. Interventions: DNA was extracted from 22 Hungarian galactosemic patients' blood (age: l mo-20y) and the GALT gene was amplified by PCR techniques. The amplified DNA was analysed for the common classic mutation causing severe galactosemia (Q188R) and the Duarte v. (N314D) and for K285N mutation. 4. Results: Q188R mut. was found in 12 out of 22 (54.54%), there were 3 compound heterozygotes for Q188R/N314D (13.63%), 6 out of 22 ones revealed to be N314D Duarte variants, (31.81%), and 8 out of 13 patients for K285N mutation (61.53%). 5. Conclusion: In the Hungarian Caucasian galactosemic patients the frequency for Q188R was 54.54%, for N314D mutation was 31.81%, and K285N mutation was 61.53%.
 the arginine is substituted for glutamine at codon 188 in exon 6, with nearly 70% prevalence in Caucasians with galactosemia. In the Duarte mutant allele of the galactose-1-phosphate uridyl- transferase GALT, EC ) GALT genome in base pair 2744 of exon 10 there is an A to G transition resulting an acidic aspartate (D) substitution for an asparagine (N) at codon 314 (N314D) (Reichardt et al. 1992). 2. Subjects: Genotypes of GALT deficient Hungarian galactosemic patients were determined as a population genetic study. 3. Interventions: DNA was extracted from 22 Hungarian galactosemic patients blood (age: l mo-20y) and the GALT gene was amplified by PCR techniques. The amplified DNA was analysed for the common classic mutation causing severe galactosemia (Q188R) and the Duarte v. (N314D) and for K285N mutation. 4. Results: Q188R mut. was found in 12 out of 22 (54.54%), there were 3 compound heterozygotes for Q188R/N314D (13.63%), 6 out of 22 ones revealed to be N314D Duarte variants, (31.81%), and 8 out of 13 patients for K285N mutation (61.53%). 5. Conclusion: In the Hungarian Caucasian galactosemic patients the frequency for Q188R was 54.54%, for N314D mutation was 31.81%, and K285N mutation was 61.53%..")
28
Family members proposita propositus mother father maternal grandmother maternal grandfather paternal grandmother paternal grandfather Age 20 y 19 y 40 y 41 y 72 y 73 y 66 y 67 y Sings II. family IV/2 IV/5 III/4 III/5 II/7 II/8 II/15 II/16 G-1 PUT U 0.3 0.5 12.1 8.6 24.3 9.1 20.4 6.7 Genotypes mutations comp heterozygote Q188R/non Q188R comp heterozygote Q188R/non Q188R obligatoric heterozygote obligatoric heterozygote heal thy heterozygote heal thy heterozygote

29
Methods DNA had been extracted from the galactosemic patients’ blood spot and the GALT gene amplified by PCR techniques. The amplified DNA was analysed for the common classic mutation causing severe galactosemia (Q 188 R) and the Duarte variant (N 314). The different genotypes have been compared to the quantitative GALT enzyme activities.
 and the Duarte variant (N 314). The different genotypes have been compared to the quantitative GALT enzyme activities..")
31
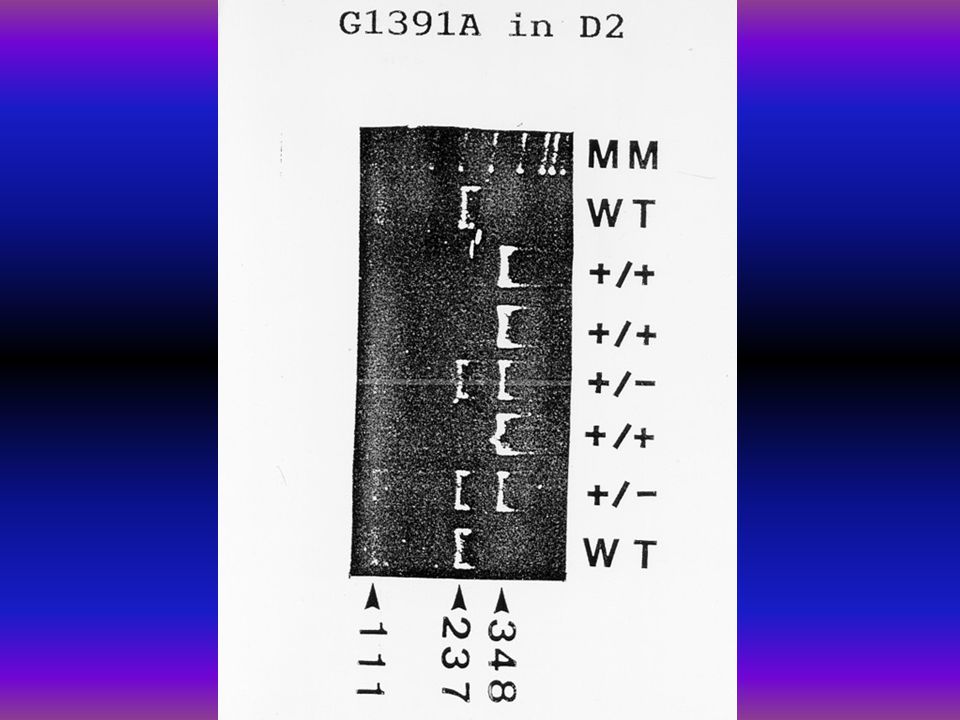
WTG 1391 A (+/+)
")
34
Genotypes and allele frequencies of the East-Hungarian galactosemic and Duarte patients' cohort n = 32 All G-1-PUT Q 188R K285 N D1 (N314D) D2 (G1391A) D1 + D2 Unknown homo- zygotes 2 0 – hetero- zygotes 12 9 4 12 16 – mutation % 60.86 39.13 20.68 34.48 55.17 13.04 allele frequency % 34.78 19.56 10.04 17.24 27.58 6.52
 D2 (G1391A) D1 + D2 Unknown homo- zygotes 2 0 – hetero- zygotes – mutation % allele frequency %")
Hasonló előadás















 7) 8) 9) 10) Mennyi az x, y és z értéke? 11) 12) 13) 14) 15)>")






