Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
Bioinformatika gyakorlat (biomérnök hallgatóknak)
Adatbázisok Szekvencia illesztés Hasonlóság keresés Filogenetikai programok PCR primer tervezés Promóter predikció (génpredikció)
")
2
Kiindulási „anyag” a szekvencia
Molekuláris biológiában, taxonómiában/filogenetikában használt „objektum” a szekvencia DNS nukleotidsorrend Fehérje aminosav-sorrend Milyen információt hordoz Gén/géntermék A szekvencia „hasonlóságban” rejlő információ Leszármazás/eredet/változás Funkcionális Szerkezeti

3
Egyéni (csoportos) feladat beadása elektronikus formában
 feladat beadása elektronikus formában")
4
Szekvencia manipuláció
Adatbázisok Szekvencia letöltés/manipuláció Hasonlóság kimutatása – számítógép szerepe > sm nt sequence GGGCGAGCGATAGTGCTAACTCAAAAAATCCTATGAGGACGAACCATGGCCAAGCGAAAGGCGCGCATCGACAGCGCCGCCGAAGCCGTGCGCGTCATGGCGAAGGCAACCCCCGAGATTGAGCCGCCGGCAAACGTGCCGCTCGACGAAGAAGACCTGCCGTTCTTCCGCAACGTGATCGCTGAGTACGCGCGGTCGGAATGGTCGTCGCATCAACTCGAGCTCGCCGCAATGCTGGCCCGCACCATGGCGGACCTGACGCGCGAGCAGAAACTGCTGCGAGACGAAGGCGGTGTTGCCTACTCCGAAAAAGGCACACCTGTCGCGAATCCGCGCAAGTCGATTGTGCAAATGCACGCCAGTTCGATCCTGTCCTTTCGTCGATCATTGTCGCTTCACGCACGCGCGCAAGCGGGCGAGGCGAGGGACGTTGCGAAGCGCC > sm nt sequence

5
> sm nt sequence CGAACCATGGCCAAGCGAAAGGCGCGCATCGACAGCGCCGCCGAAGCCG TGCGCGTCATGGCGAAGGCAACCCCCGAGATTGAGCCGCCGGCAAACGTG CCGCTCGACGAAGAAGACCTGCCGTTCTTCCGCAACGTGATCGCTGAGTAC GCGCGGTCGGAATGGTCGTCGCATCAACTCGAGCTCGCCGCAATGCTGGC CCGCACCATGGCGGACCTGACGCGCGAGCAGAAACTGCTGCGAGACGAA GGCGGTGTTGCCTACTCCGAAAAAGGCACACCTGTCGCGAATCCGCGCAA GTCGATTGTGCAAATGCACGCCAGTTCGATCCTGTCCTTTCGTCGATCATTG TCGCTTCACGCACGCGCGCAAGCGGGCGAGGCGAGGGACGTTGCGAAGC GCC > sm nt sequence GGGCGAGCGATAGTGCTAACTCAAAAAATCCTATGAGGACGAACCATGGCC AAGCGAAAGGCGCGCATCGACAGCGCCGCCGAAGCCGTGCGCGTCATGG CGAAGGCAACCCCCGAGATTGAGCCGCCGGCAAACGTGCCGCTCGACGA AGAAGACCTGCCGTTCTTCCGCAACGTGATCGCTGAGTACGCGCGGTCGG AATGGTCGTCGCATCAACTCGAGCTCGCCGCAATGCTGGCCCGCACCATGG CGGACCTGACGCGCGAGCAGAAACTGCTGCGAGACGAAGGCGGTGTTGC CTACTCCGAAAAAGGCACACCTGTCGCGAATCCGCGCAAGTCGATTGTGCA AATGCACGCCAGTTCGATCCTGTCCTTTCGTCGATCATTGTCGCTTCACGCA CGCGCGCAAGCGGGCGAGGCGAGGGACGTTGCGAAGCGCCGGGCGAGC GATAGTGCTAACTCAAAAAATCCTATGAGGA

6
Illesztett szekvencia
CGAACCATGGCCAAGCGAAAG ||||||||||||||||||||| GGGCGAGCGATAGTGCTAACTCAAAAAATCCTATGAGGACGAACCATGGCCAAGCGAAAG GCGCGCATCGACAGCGCCGCCGAAGCCGTGCGCGTCATG |||||||||||||||||||||||||||||||||||||||.... GGGACGTTGCGAAGCGCC ....|||||||||||||||||| GGGACGTTGCGAAGCGCCGGGCGAGCGATAGTGCTAACTCAAAAAATCCTATGAGGA ||||||||||||||||||||||||||||||||||||||| GGGCGAGCGATAGTGCTAACTCAAAAAATCCTATGAGGA

7
Adatbázisok NCBI, EBI, GenomeNet PubMed Elsődleges (nukleotid)
Származtatott (aminosav szekvencia, egyéb jellemzők

8
Illesztési módszerek a hasonlóság függvényében
A skála az illesztett fehérjeszekvenciák közötti százalékos egyezést mutatja Két véletlen szekvencia illesztése ~20 %egyezést mutat 20 % alatt nem szignifikáns az illesztés

9
Homológia és hasonlóság
A homológia fogalmát gyakran hibásan használják Két szekvencia homológ, ha közös őstől származik Az analóg szekvenciák olyan nem homológ szekvenciák, amelyekben hasonló szerkezetű részek vagy hasonló funkcionális helyek találhatók, és ezek konvergens evolúcióval jöttek létre A homológia nem a hasonlóság mértéke Az olyan kifejezések, mint „a szekvenciák 50 %-banhomológok” vagy „a szekvenciák nagyfokú homológiát mutatnak”, értelmetlenek A hasonlóság egy tény, a homológia egy hipotézis ill. következtetés A hasonlóság kvantitatív, a homológia kvalitatív

10
Alapfogalmak Szekvencia illesztés ->Homológia-vizsgálat kimutathatja: Leszármazási viszonyokat Szerkezetet illetve funkciót mutathat Szekvencia hasonlóság, szerkezeti és ezáltal funkcionális hasonlóságot mutat

11
Szignifikancia A matematikai és a biológiai szignifikancia különbözik
Pl. kis komplexitású régiók, konvergens evolúcióval létrejött hasonlóság Ebből fakadó korlátok Az adatbázis-kereső algoritmusokban A szekvenciaillesztő algoritmusokban A mintázat-felismerési módszerekben A funkcionális hely-és szerkezetpredikciós eszközökben Ezért mindig ajánlatos a szekvenciaelemző módszerek széles körét alkalmazni Egyik módszer sem tévedhetetlen!

12
Szekvencia illesztés során az evolúciós változások figyelembe vehetők
Pl. ha van 2 rokon fehérje, ugyanaz a funkciója Ha egymáshoz illesztjük a szekvenciát sok eltérés lehet Ettől még hasonló A helyettesítési mátrixok ezt veszik figyelembe

13
Változások az evolúció során
Nukleotid sorrendben Pontmutáció: tranzíció, transzverzió Nukleotid szubsztitúció: pontmutáció SNP – single nucleotide polymorphism Szegmentális mutáció: del, in, inv – ha nem egy nukleotidot érint Szinonim ill. nem-szinonim szubsztitúció - vigyázat! – nem biztos, hogy a szinonim szubsztitúció kihatás nélküli – pl. splicing megváltoztatása Nukleotidok nem-random előfordulása: a 4 bázist tartalmazó nukleotidok nem 1:1:1:1 arányban találhatók a genomokban; Dinukleotidok nem-random előfordulása: funkcionális kényszerek a genetikai kód miatt

14
Nukleotid sorrendben (folyt.)
Nem-random a szinonim kodonok használata: RSCU – relative synonymous codon usage n = a szinonim kodonok száma egy aminosavra (1 – 6) i = egy adott kodon Xi= egy adott kodon előfordulási száma ha azonos gyakorisággal használtak, akkor az RSCU-k összege 1 Effektív kodonszám: ENC 20 – 61: 20 – minden aminosavra egy kód 61 – teljesen random, nincs kiemelt kód
 i = egy adott kodon. Xi= egy adott kodon előfordulási száma. ha azonos gyakorisággal használtak, akkor az RSCU-k összege 1. Effektív kodonszám: ENC. 20 – 61: 20 – minden aminosavra egy kód. 61 – teljesen random, nincs kiemelt kód.")
15
Helyettesítési modellek (nukleinsav)
Kodonhasználat: nem teljesen univerzális a genetikai kód: mitochondriális genom, néhány prokarióta, egysejtű, ill. gomba spec. kodonokkal rendelkezik hiányzó kodonok: néhány szervezetben a fehérjéket kódoló régiókban sosem fordulnak elő bizonyos kodonok hozzá nem rendelt kodonok: a kodon megtalálható, de nincs hozzá tRNS a megfelelő antikodonnal – leáll a transzláció, a polipeptid a riboszómához kötötten marad Azonossági (szubtitúciós) mátrix nukleotidokra Pl: egyezés 1, mismatch 0 (vagy egyezés 5, eltérés -4) Esetleg súlyozható Tranzíció kisebb súllyal Transzverzió nagyobb súllyal (előadás későbbi részében)
 mátrix nukleotidokra. Pl: egyezés 1, mismatch 0 (vagy egyezés 5, eltérés -4) Esetleg súlyozható. Tranzíció kisebb súllyal. Transzverzió nagyobb súllyal (előadás későbbi részében)")
16
Szekvencia változások az evolúció során
Aminosav/fehérje szinten Aminosav kémiai tulajdonságait, hasonlóságait figyelembe vevő márix A hasonló fiziko-kémiai tulajdonságokkal rendelkező aminosavak illeszkedését nagyobb súllyal veszi figyelembe (osztályozás alapja: poláros vagy apoláros, méret, alak, töltés) Észlelt helyettesítéseken alapuló szubsztitúciós mátrixok Az illesztett szekvenciákban észlelt aminosav-helyettesítési gyakoriságokon alapulnak Fehérjeszekvenciák illesztésénél ma már szinte csak ilyeneket használnak Pl: PAM, BLOSUM
 Észlelt helyettesítéseken alapuló szubsztitúciós mátrixok. Az illesztett szekvenciákban észlelt aminosav-helyettesítési gyakoriságokon alapulnak. Fehérjeszekvenciák illesztésénél ma már szinte csak ilyeneket használnak. Pl: PAM, BLOSUM.")
17
Észlelt helyettesítéseken alapuló mátrix
Dayhoff mutációs mátrix Közeli rokon szekvenciák illesztéséből (legalább 85% hasonlóság, 70 illesztés, helyettesítés megfigyeléséből) PAM mátrix létrehozása (log odds mátrix) PAM = Percent Accepted Mutation 1 PAM az adott evolúciós időtartam alatt 1 aminosav kicserélődése elfogadott (accepted) 100 aminosav esetén (1%) Feltételezés: a mutáció független az aminosavtól (hogy mi volt előtte) és a pozíciótól (hol helyezkedik el a szekvenciában) A fenti alapján interpolálható az aminosavak változása PAM1xPAM1=PAM2 (kétszer olyan távoli esemény esetén az aminosav változás) PAM250 (PAM1250) kb. 20% aminosav azonosságot jelent
 PAM mátrix létrehozása (log odds mátrix) PAM = Percent Accepted Mutation. 1 PAM az adott evolúciós időtartam alatt 1 aminosav kicserélődése elfogadott (accepted) 100 aminosav esetén (1%) Feltételezés: a mutáció független az aminosavtól (hogy mi volt előtte) és a pozíciótól (hol helyezkedik el a szekvenciában) A fenti alapján interpolálható az aminosavak változása PAM1xPAM1=PAM2 (kétszer olyan távoli esemény esetén az aminosav változás) PAM250 (PAM1250) kb. 20% aminosav azonosságot jelent.")
18
Helyettesítési mátrixok
BLOSUM mátrixok BLOCKS adatbázisból származó adatokból Távolabbi rokon szekevenciák hézag nélküli blokkjainak többszörös illesztése BLOcks SUbtitution Matrix BLOSUM Log odds mátrix

19
BLOCKS adatbázis —> BLOSUM
Többszörös illesztés -> blokkok Rokon szekvenciák hézag nélküli blokkjainak többszörös illesztése Nincsenek hézagok, konzervált régiók -> megbízhatóbb illesztése Klaszterezés páronkénti hasonlóság alapján pl. minden szekvencia 80% hasonlóságot mutat minden másikkal Ebből helyettesítési gyakoriságok számolása -> mátrix Pl: BLOSUM80

20
Hogyan készül a mátrix Általános képlet (log odds)
Sij pontérték mátrixelem (Score az adott mátrix elemre, negatív várható pontérték random szekvenciákra) λ=pozitív konstans qij észlelt aminosavpár gyakoriság az illesztésekben (célgyakoriság, ∑=1) pi és pj aminosav előfordulási gyakoriságok az adott aminosavra (háttérgyakoriságok)
 λ=pozitív konstans. qij észlelt aminosavpár gyakoriság az illesztésekben (célgyakoriság, ∑=1) pi és pj aminosav előfordulási gyakoriságok az adott aminosavra (háttérgyakoriságok)")
21
BLOSUM62 mátrix

22
Páronkénti összehasonlítás
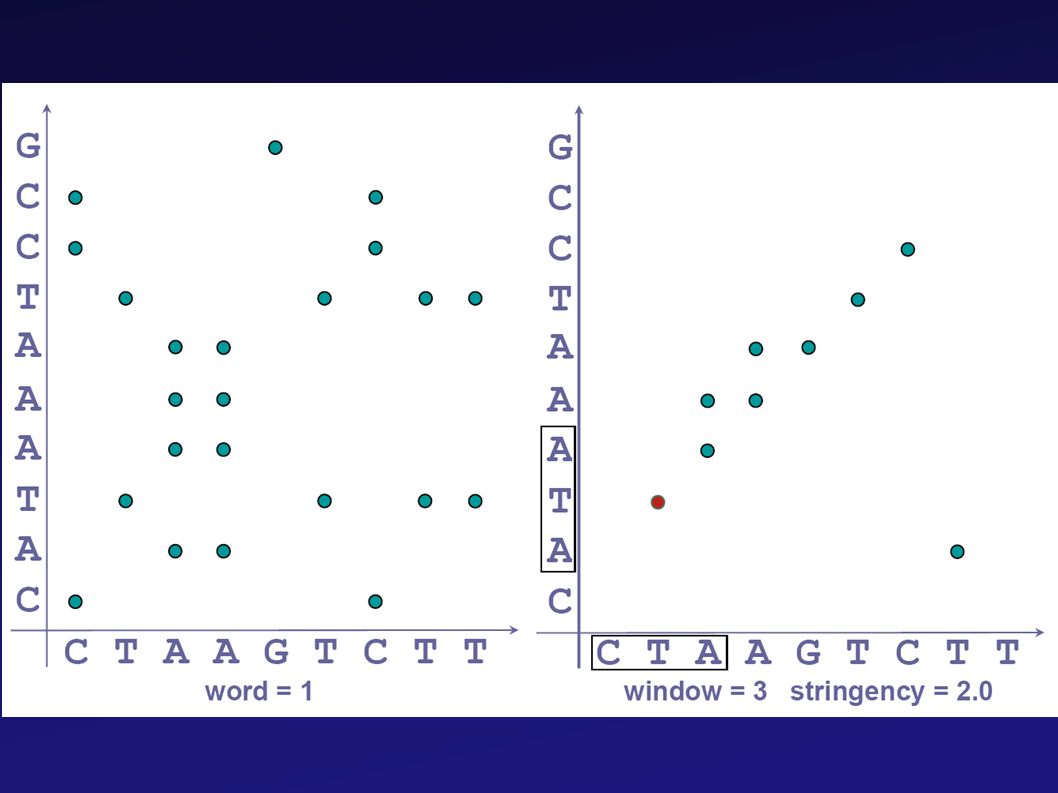
Pontábrázolás (dotplot)
")
24
A dotplotok értelmezése

25
Fehérje dotplot

26
Fehérje dotplot

27
Nukleinsav dotplot

28
Illesztés pontozása Helyettesítési mátrixból vett pontértékek
Pl: PAM 250 helyettesítési mátrix felhasználásával 1. szekv. M N A L S D R T 2. szekv. M S D R T T E T pont = 7

29
BLOSUM62 helyettesítési mátrix

30
Páronkénti illesztések
Optimális illesztések: Globális és lokális illesztés

31
„Optimális” illesztések
Szekvencia-illesztés(„alignment”): két szekvencia olyan elrendezése, amely megmutatja, hogy a két szekvencia hol hasonlít, illetve hol különbözik egymástól hipotézis: pozícionális homológia Optimális illesztés: a legtöbb egyezést és a legkevesebb különbséget mutató szekvenciaillesztés Matematikailag optimális, azaz az alkalmazott pontozási rendszerrel a legnagyobb pontértéket mutató illesztés Hogy egy adott esetben a nagyszámú lehetséges illesztés közül melyik lesz optimális, az nagymértékben függ az alkalmazott pontozási módszertől!
: két szekvencia olyan elrendezése, amely megmutatja, hogy a két szekvencia hol hasonlít, illetve hol különbözik egymástól. hipotézis: pozícionális homológia. Optimális illesztés: a legtöbb egyezést és a legkevesebb különbséget mutató szekvenciaillesztés. Matematikailag optimális, azaz az alkalmazott pontozási rendszerrel a legnagyobb pontértéket mutató illesztés. Hogy egy adott esetben a nagyszámú lehetséges illesztés közül melyik lesz optimális, az nagymértékben függ az alkalmazott pontozási módszertől!")
32
Páronkénti illesztés: lokális vagy globális

33
Hézagok pontozása (gap penalty)
Lehet fix, de általában a hézag hosszával növekszik „Affin” hézagbüntetés (wx, két részből áll): Hézagnyitási büntetés (nagy): g Hézagkiterjesztési büntetés (kisebb): rx wx=g+rx ahol x a hézag hosszúsága A pontozási mátrixtól függetlenül is módosíthatók, de a pontozási mátrix korlátozza, hogy milyen határok között Az alapértelmezett értékek általában tapasztalati úton lettek beállítva, és tipikus feladatokra jól használhatók, pl. BLAST-nál
: Hézagnyitási büntetés (nagy): g. Hézagkiterjesztési büntetés (kisebb): rx. wx=g+rx ahol x a hézag hosszúsága. A pontozási mátrixtól függetlenül is módosíthatók, de a pontozási mátrix korlátozza, hogy milyen határok között. Az alapértelmezett értékek általában tapasztalati úton lettek beállítva, és tipikus feladatokra jól használhatók, pl. BLAST-nál.")
34
Páronkénti illesztés algoritmusa

35
Az illesztési mátrix kitöltése (Hij értékek)
")
36
Az illesztések eredménye
Két alternatív globális illesztés sequence 1 M - N A L S D R T sequence 2 M G S D R T T E T Score = -5 sequence 1 M N - A L S D R T Score = -5 Globális illesztés, de nincs hézagbüntetés a végeken sequence 1 M N A L S D R T Sequence M G S D R T T E T Score = 10

37
Többszörös illesztés Multiple sequence alignment (MSA vagy msa)
Kettőnél több szekvencia optimális illesztése Célja hogy minél több egyező karaktert (nukleotid, vagy AA) egy oszlopba rendezzen Tartalmazhat illeszkedést (match), nem illeszkedést (mismatch) és hézagokat (gap) (emiatt bonyolult probléma
 egy oszlopba rendezzen. Tartalmazhat illeszkedést (match), nem illeszkedést (mismatch) és hézagokat (gap) (emiatt bonyolult probléma.")
38
Többszörös illesztési megközelítések
Dinamikus programozás kiterjesztése Progresszív globális illesztés Iteratív módszer Motívumokat felhasználó illesztés Statisztikai módszerek probabilisztikus modellek felállításával

39
A többszörös illesztésben lévő információ
Egy adott szekvencia készlet többszörös illesztése úgy tekinthető, mint a Szekvenciák evolúciós töténete Azok a szekvenciák, amelyek jól illeszkednek, valószínűleg később divergálódtak a közös ős szekvenciától Olyan szekvencia csoport, amelyik rosszabbul illeszthető, komplexebb és távolibb evolúciós kapcsolatot mutat

40
Az illesztés megvalósítása egyenértékű a szekvenciák közötti evolúciós kapcsolatok feltárásával

41
Többszörös illesztés Nehéz vagy bonyolult feladat?
Nagy hasonlóság esetén triviális Inzerciók, deléciók esetén nem hogy nem triviális, hanem még számítógépekkel is bonyolult feladat

42
Felhasználás Szekvenálás (genom szekvenálás, shotgun szekvenálás)
Strukturális funkcionális részek azonosítása Fehérjében, domén vagy katalitikus aminosav Nukleotid szekvenciában pl: promóter fehérje kötő hely stb.

43
Felhasználás Új szekvenciák és meglévő családok közötti hasonlóság (és homológia) felderítése Másodlagos és harmadlagos fehérje szerkezetek előrejelzésének segítése (pl. homológia modellezés) Oligonukleotid primerek tervezése Filogenetikai analízis alapfeltétele
 Oligonukleotid primerek tervezése. Filogenetikai analízis alapfeltétele.")
44
MSA és evolúciós fa kapcsolata
Optimális illesztés minimalizálja a fán a mutációs lépések számát

45
MSA dinamikus programozási algoritmussal
Probléma 2 szekvencia összehasonlításánál Az összehasonlítások száma NxM, ahol N az egyik szekvencia hossza, míg M a másiké Tegyük fel, hogy a két szekvencia egyforma hosszú, N=M, ekkor az összehasonlítások száma N2 10 szekvencia esetén az összehasonlítások száma N10 pl. 10db 300 AA hosszúságú fehérje esetén 30010=5,9x1024

46
Scoring mátrix 3 szekvenciára
A szekvencia A-B B szekvencia A-C MSA (A-B-C) B-C C szekvencia
 B-C. C szekvencia.")
47
Egyszerűsítés

48
Progresszív módszer ClustalW (a leggyakrabban használt program)
1. minden szekvencia páros összehasonlítása 2. az illesztési pontértékek felhasználása filogenetikus fa készítéséhez (vezérfa) 3. egymás utáni szekvencia illesztés a vezérfa alapján Először a leghasonlóbb szekvenciák illesztése történik meg, majd ehhez illeszti az egyre kevésbé hasonlító szekvenciákat
 3. egymás utáni szekvencia illesztés a vezérfa alapján. Először a leghasonlóbb szekvenciák illesztése történik meg, majd ehhez illeszti az egyre kevésbé hasonlító szekvenciákat.")
49
Clustal Távolságmátrix a páros illesztésekből Vezérfa szerkesztése
Illesztés a legnagyobb hasonlóságot mutató szekvenciák illesztésével

50
Legfőbb probléma a progresszív illesztéssel, hogy a kezdeti illesztéstől nagymértékben függ a többszörös illesztés Ha hasonló szekvenciákra nézzük, akkor jó eredmény Az elején beillesztett gap pl. nem módosul az illesztés során vagy a korán elkövetett illesztési probléma kihat az egész illesztésre (lokális minimum probléma, az algoritmus „greedy” mohó természetéből fakad

51
Iteratív módszerek pl. MultAlign
Újraszámolja a páros illesztések pontértékeit a progresszív illesztés során Az újraszámolt pontérték alapján új fát készít Az új fa alapján javítja az illesztést

52
Genetikus algoritmus Alapötlet
Sokféle illesztést generálunk átrendeződésekkel, és rés beépítésével egy egy generáció során Az utódok (illesztések) közül kiválasztjuk legjobb pontértéket adót
 közül kiválasztjuk legjobb pontértéket adót.")
53
Profil alapú illesztés
Helyi hasonlóságok felhasználása az illesztésekben Profil analízis Kisebb hasonló (nagymértékben konzerválódott) darabokat illeszt (nem globális) A hasonló darabok alapján profilt (egyfajta pontozási mátrixot képez) Ebben benne van az aminosavak helyettesítési értéke is Réseket is magában foglalja
 darabokat illeszt (nem globális) A hasonló darabok alapján profilt (egyfajta pontozási mátrixot képez) Ebben benne van az aminosavak helyettesítési értéke is. Réseket is magában foglalja.")
54
Statisztikai módszerek
Rejtett Markov Láncok

55
Statisztikai módszerek
Rejtett Markov Modell (Hidden Markov Model, HMM)
")
56
HMM Valószínűségelméleti leírása a szekvencia illesztésnek
Statisztikai modell Minden lehetséges illeszkedő/nem illeszkedő pozíciót és rést figyelembe vesz az msa generálásához A szekvencia családból modell készül (előzetes/priori információból kiindulva darabos szekvencia készlet használható a modell tanítására A tanított modell használható az msa létrehozására (posterior információ)
")
57
Hasonlóság keresés A probléma:
Van egy szekvenciánk, amiről nem tudunk semmit Van az adatbázis, ahol a már ismert/jellemzett szekvenciákat letárolták Hasonlítsuk össze a „saját” szekvenciát az adatbázisban találhatókkal Ha szekvencia egyezést, vagy hasonlóságot találunk, akkor a funkcióra következtethetünk

58
A korábbi illesztés (Smith-Waterman, Nedleman-Wuntsch) nagyon jó, de lassú, ha nagy adatbázisok vannak Heurisztikus megoldások (nem minden illesztés jön ki, de gyors)
")
59
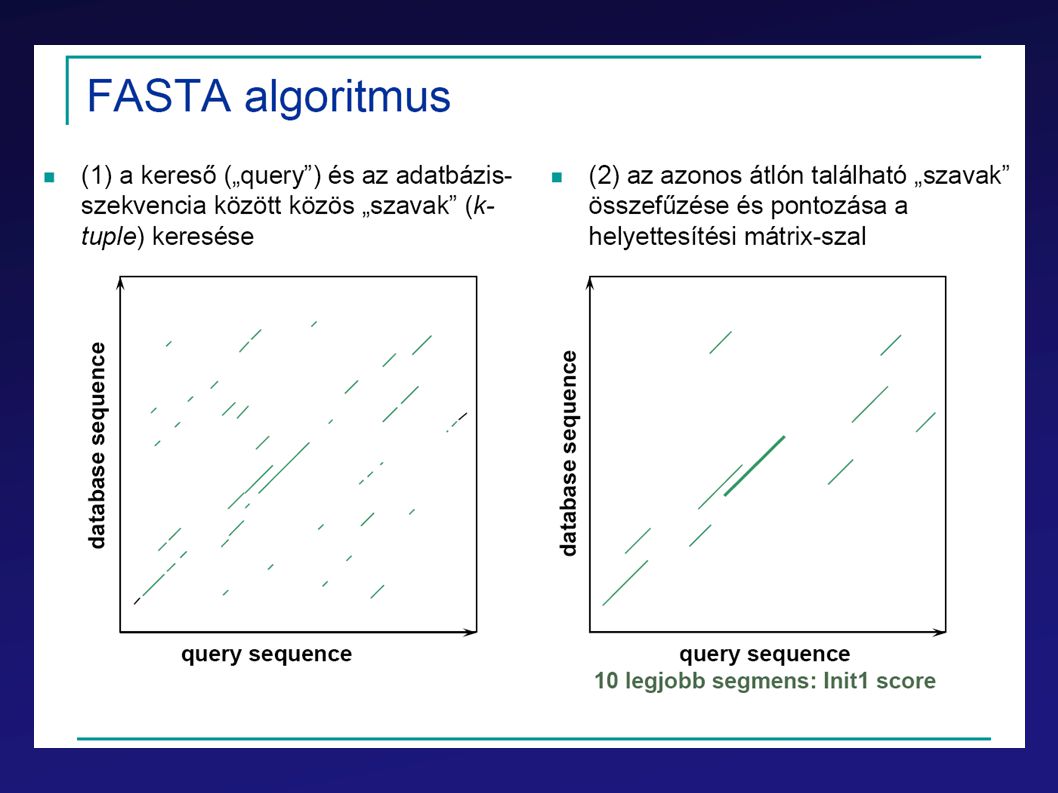
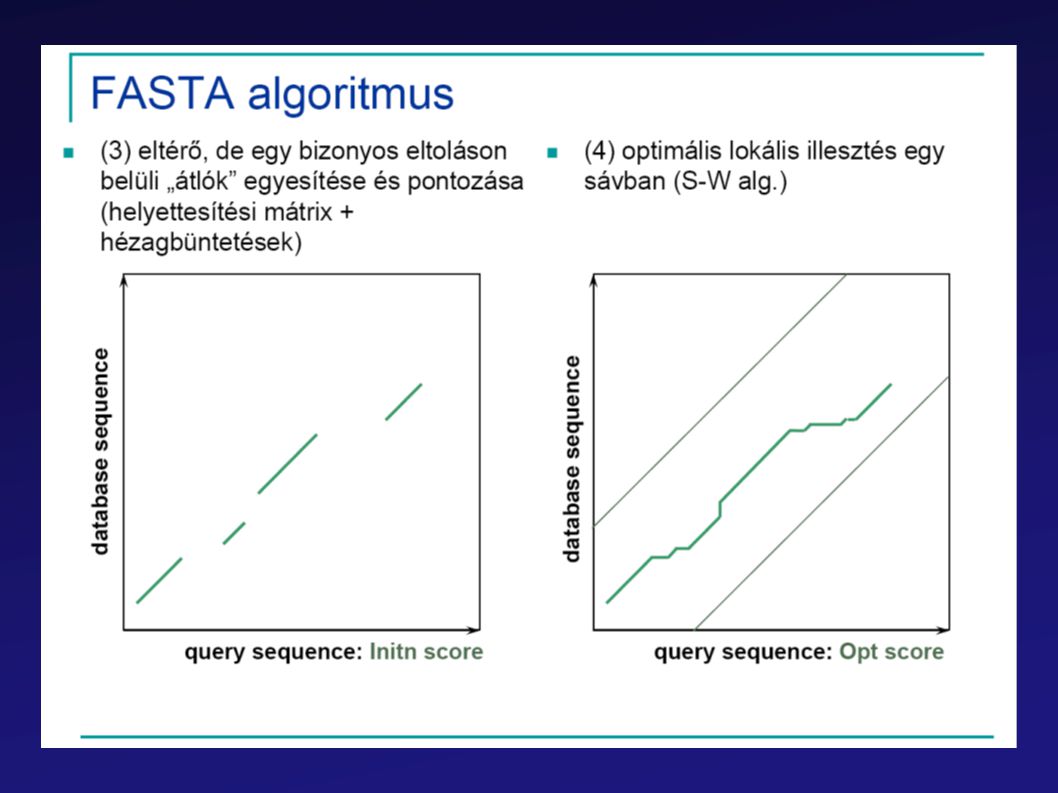
FastA BLAST PSI-BLAST

62
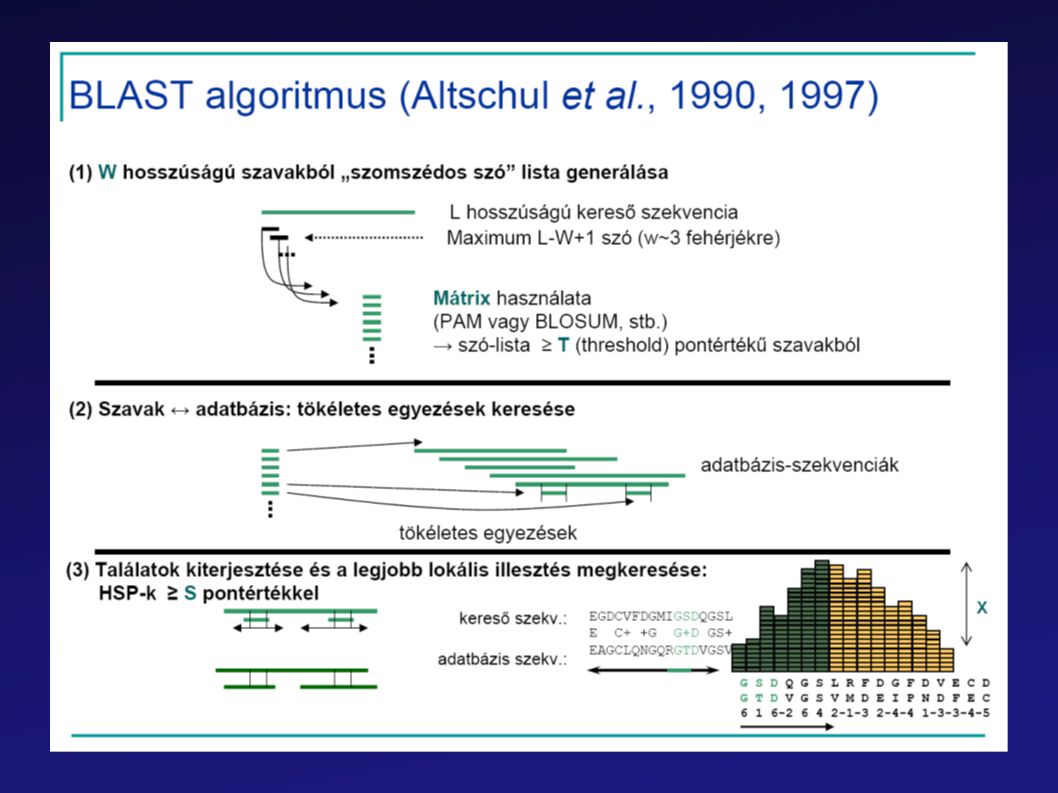
BLAST Leggyorsabb (akár helyi gépeken is használható)
Gyors lokális illesztéseket végez Statisztikai módszerek alkalmazásával becsüli a találatok szignifikanciáját

64
Statisztikai szignifikanciabecslés:
E érték: hasonló vagy nagyobb pontértékű találat véletlen előfordulásának várható száma; minél kisebb, annál jobb.

65
Promóter predikció

66
Feladatok: 1. Feladat: PubMed keresés
Hány xilanáz-ról (növényi sejtfalbontó enzim, xyalanase) szóló publikáció van a PubMed adatbázisban? Mennyi jelent meg 2000-ben? Ebből hány publikációban szerepelt Gilbert nevezetű szerző?
 szóló publikáció van a PubMed adatbázisban Mennyi jelent meg 2000-ben Ebből hány publikációban szerepelt Gilbert nevezetű szerző")
67
2. Feladat: BLAST keresés
CGCATCGTCGGCATATGGTTCGGTGAGCGCTACCCTGGCGCACTTATTGGACTTCCGACGGGCGAGCCTCTTGGCGCA TGGGTTCTCGACCTTGACCGACATGGCGATCGTGACGGGCATGCGTGGCTCGCCGAGATGGAGGCGAAGCACGGCG CGCTGCCCGAAACAGCAAGAGCCAGCACGGCCAACGGTGGAACGCACATATTCTTCAAGCACGTCGCCGGCATTCGC AACCGTGCGGCAATCGCGCCTGGTGTGGATACTCGAGGAGATGGCGGCTATGTCTGCGGGCCTGGCTCGCAAATGGC CGATGGCCGAAGATACCAGTGGATCGACTACGATGGAGACGGGCTGCCTCCCATCGCGGATGCCCCTGCATGGCTCAT CGACCTGCTGAAGCCGAAGGTGGTGGAAGCCGCCGAAAGGCGCCAGCCGTCCACATACACATACCAGCCGGAGGATA GCGGCGCTGCTCGCTATGCCGCCAAGTCCTTTGAAATGGAGCTCGAGAAGCTGCGCAACTCGCCCAGCGGCCAGCGC GGTCAGCAGTTGTTCGCCAGCGCGTGCTCCATCGGTGAGTTCGTTGCCGGAGGCCTGATTTCACGATCGGAGGCCGA GGCTGGTCTGCTGGACGCGGCAGCGGCGTGCGGCGTGCTCCAGAAGGACGGCGAGCGAAAGACTGTGGACCGTATC CGGCGAGGGCTGGACAAGACGGCGAACACGCCGCGGCAGATACCGGAGCGCGAATATGACAACGACAACACGCCAG TAAACGCTGCGGAAATGGAGGCTTTCGTCGAGCGGCACAAGGCCAAGAAGGAGGCGGCAGCGCAGCAACAAACCGC CGTTGTTGAGGAGCAGCCACCAGCCGACCAGGCACCGACGCCCCGCCAGAAGGCCCGATTCGAACTGACGTGGTTC GATGACATCGAGGAGGGCAAGCCGAAGGAGACCATCCTCAAGGGCTGGCTAGGCGTTGGCGAATTCACCACCATCTC GGGCCTGCCGGGAACTGGCAAGAGCGTCGTGACAACCGACCTGGCTTGCCACATCGCGGCTGGCATGGACTGGCAT GGCATGAAGGTCCAGCAGGGCCTGGTTGTCTACGTGGCGGCCGAGCGCAAGAAGCTGACGGAGCGGCGCATGATGG CCTTCCGCAAGCACCATAACAAACACAACGTGCCCCTTCTCGTCGTAGGCGGCATGCTGGACTTTACCCGCGATCTGAA GGACGCCGAGGACATAATCAAGGTGATCAGGGAGGCGGAAACCATCACAGGCATGAAGTGCGTGTGGGTCATTATCGA CACGCTCACCCGCACATTCGGAGCCGGCGACCAGAACGCATCCAAGGATATGGTGAAGTTCGTTCGATCCTGCGACAA AATCGTAGAGGACATCGGGGCGCATGTTACGGCTATCCACCATTCATCGTGGAACGGTGAGCGAGGGAAGGGCGCCAT CGACCTGGACGGGGCAGTCGATGCTTCATTCATGGTGAAGAAGGACGGCAACAAGCACAGACTCGTCTGCGACGGGA CCAACGACGGAGAGGATGGCGACGTGCTGGCCTTCACCATGCAATCCGTT Mi lehet a funkciója a következő szekvenciának? Miből származhat a szekvencia nagy valószínűséggel?

68
3. Feladat: Primer tervezés
Az adatbázisból keresse ki az Azotobacter vinelandii ANFH (alternatív nitrogén fixálás) génjét Töltse le a szekvenciát Keresse ki a Chlamydomonas reinhardtii aktin génjét (actin) Tervezzen primer párt az ANFH génre, ami 450 bp hosszúságú szakaszt amplifikál. Tervezzen primer párt az actin génre, amely 525 bp hosszúságú szakaszt amplifikál. Mire kell figyelni a Chlamydomonas primer tervezésénél? (Eukarióta szervezet!)
 génjét. Töltse le a szekvenciát. Keresse ki a Chlamydomonas reinhardtii aktin génjét (actin) Tervezzen primer párt az ANFH génre, ami 450 bp hosszúságú szakaszt amplifikál. Tervezzen primer párt az actin génre, amely 525 bp hosszúságú szakaszt amplifikál. Mire kell figyelni a Chlamydomonas primer tervezésénél (Eukarióta szervezet!)")
Hasonló előadás

 DNS-ből,>")












>")


.>")

 Csernetics Árpád Bioinformatika SZIT 2005. ápr. 18.>")