Előadást letölteni
1
Bioinformatika Szekvenciák és biológiai funkciók ill. genotipusok és fenotipusok egymáshoz rendelése Kós Péter 2009.XI.

2
Egyes molekulák hasonlósága
A BLAST korlátai A BLAST tár- ill. időigénye O (n x m) Nagyon hosszú szekvenciák (teljes genomok) összehasonlítására nem alkalmas Figyelembe veszi a vizsgált szekvenciák minden elemét Az egyes régiók súlyozására nincs lehetőség, így az esetleg „felhígult” információt nem találja meg 1 2 Genomok, kromoszómák Egyes molekulák hasonlósága Aktív helyek, molekula részek BLAST, FASTA
 Nagyon hosszú szekvenciák (teljes genomok) összehasonlítására nem alkalmas. Figyelembe veszi a vizsgált szekvenciák minden elemét. Az egyes régiók súlyozására nincs lehetőség, így az esetleg „felhígult információt nem találja meg Genomok, kromoszómák. Egyes molekulák hasonlósága. Aktív helyek, molekula részek. BLAST, FASTA.")
3
Összehasonlító genomika
A genomok géntartalma, szerveződése rengeteg információval szolgál gén a b c d e + - genom

4
Genomok összehasonlítása: MegaBLAST
„Fösvény algoritmus” (Greedy algorithm) csak ott használ dinamikus programozást, ahol az elkerülhetetlen Összefűzött kérdő szekvenciák a keresést egyszerre végzi, majd az eredményből kiválogatja az egyes szekvenciákra vonatkozó adatokat
 csak ott használ dinamikus programozást, ahol az elkerülhetetlen. Összefűzött kérdő szekvenciák. a keresést egyszerre végzi, majd az eredményből kiválogatja az egyes szekvenciákra vonatkozó adatokat.")
5
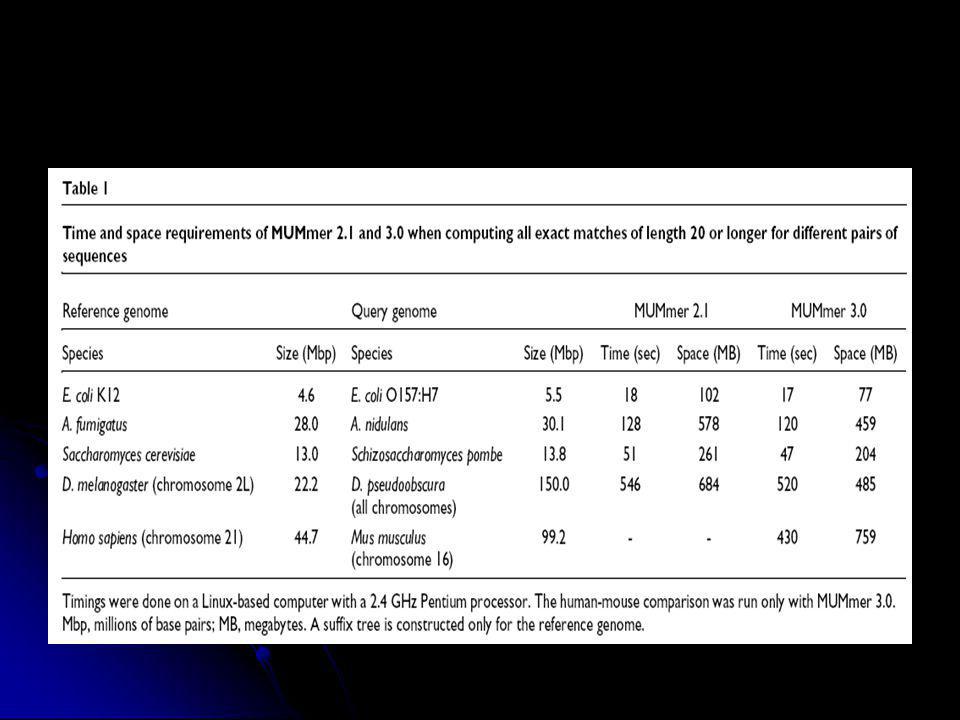
Genomok összehasonlítása: MUMmer
A szekvencia ábrázolása toldalékfa (suffix tree) formájában uvw : u-prefix (előtag), v-szöveg, w-suffix (toldalék) O (n) időigény Maximal Unique Matches (MUM) meghatározása Szomszédos MUM-ok összekötése MUMer2 : Streaming query : 1 fa + sok kis kérdés sebesség, genom szekvenálás Nucmer, prommer (nem 100%-s azonosság megtalálása) MUMmer3 Tetszőleges ABC miniproteome javított nucmer, prommer, grafikus interfész
 formájában. uvw : u-prefix (előtag), v-szöveg, w-suffix (toldalék) O (n) időigény. Maximal Unique Matches (MUM) meghatározása. Szomszédos MUM-ok összekötése. MUMer2 : Streaming query : 1 fa + sok kis kérdés sebesség, genom szekvenálás. Nucmer, prommer (nem 100%-s azonosság megtalálása) MUMmer3. Tetszőleges ABC miniproteome. javított nucmer, prommer, grafikus interfész.")
6
Genomok összehasonlítása: MUMmer
Delcher et al, NAR v. 27

7
Az agcgacgag toldalékfájanak felépítése

8
MUMmer2: 1 suffix tree, streaming query

9
Genomok összehasonlítása a MUM meghatározás után
5: Transzpozíció 3: Véletlen illeszkedés 6: MUM meghosszabbítás

11
genomok összehasonlítása MUMmerrel
Fasta 25-mers MUMmer

12
genomok összehasonlítása promerrel
nucmer promer

13
2. probléma: Egyes esetekben a rokon molekulák szekvenciájának csak egy része mutat homológiát. Ilyenkor a teljes szekvenciára kiterjedő homológia-keresés hibás eredményeket szolgáltathat

14
Mikor tekinjük szignifikánsnak a homológiát?
Mi a teendő nagy evolúciós távolságok esetén? Ekkor azonos funkció mellett is alacsony szintű a homológia. Mikor tekinjük szignifikánsnak a homológiát? E() Hasonlóság mértéke: % azonos aminosavak Mikor mondhatjuk, hogy az adott pontszám, %-os hasonlóság, vagy egyéb matematikai jellemző biológiai jelentőséggel bír?
 Hasonlóság mértéke: % azonos aminosavak. Mikor mondhatjuk, hogy az adott pontszám, %-os hasonlóság, vagy egyéb matematikai jellemző biológiai jelentőséggel bír")
15
További információk bevonása
The Twilight Zone Elvileg 2 tetszőleges AA szekvencia minden 20 aminosava „passzol”: % azonosság A gyakorlatban, az aminosavak különböző gyakorisága következtében „minden-mindennel” átlag 8%-ban azonos: Midnight Zone kb % AA azonosság mellett a szerkezetek ált. hasonlóak (backbone rms<1Å): rokon funkciók homológ szekvenciák (közös ős) ~25% aminosav azonosság alatt: a “true positive” és „false positive” találatok különválasztása lehetetlen a közös ős nemigen határozható meg puszán szekvencia-adatok alapján: Twilight Zone Segítség: Szakértői módszerek Automatizálható módszerek További információk bevonása
: rokon funkciók homológ szekvenciák (közös ős) ~25% aminosav azonosság alatt: a true positive és „false positive találatok különválasztása lehetetlen. a közös ős nemigen határozható meg puszán szekvencia-adatok alapján: Twilight Zone. Segítség: Szakértői módszerek. Automatizálható módszerek. További információk bevonása.")
16
„Több hasonló mint azonos”
Automatizálható módszerek alacsony homológiájú fehérje-párok közül a „false nagativ”-ok elvetésére „Több hasonló mint azonos” „Sequence-space-hopping”

17
Az evolúció során csökkenő szekvencia-homológia nem egyenletesen oszlik el a molekulában
Aktív molekula: 3D Részei: Aktív hely(ek) TÉRBEN közeli aminosavak Minden számít Szerkezeti elemek Az egyes aktív helyeket megfelelő pozícióban tartják (α, β, C-C) A szerkezet számít Egyéb Szelekciós nyomás konzerváltság
 TÉRBEN közeli aminosavak Minden számít. Szerkezeti elemek. Az egyes aktív helyeket megfelelő pozícióban tartják (α, β, C-C) A szerkezet számít. Egyéb. Szelekciós nyomás konzerváltság.")
18
Máshol jelentős különbségek
Pontosan illeszkedő, AZONOS aminosavak az aktív helyen A szerkezet megtartását eredményező esetleges mutációk Máshol jelentős különbségek

19
Többszintű megoldások Egyenként, vagy integráltan
Kb 2D A másodlagos szerkezet előrejelzése: alfa, beta, ACC, TM, ... PHD..., Jpred, Threader 3D Harmadlagos szerkezetek hasonlósága DALI/FSSP PROCAT ( A database of 3D enzyme active site templates ) SCOP (Structural Classification of Proteins) CATH (Class, Architectre, Topology and Homologous superfamily) CDART (protein homology by domain architecture ) Továbbra is 1D Látható, vagy láthatatlan motívumok felkutatása a primer szekvenciában profile, Pfam/Rfam, BLOCKS, CDD, COD HMM
 SCOP (Structural Classification of Proteins) CATH (Class, Architectre, Topology and Homologous superfamily) CDART (protein homology by domain architecture ) Továbbra is 1D. Látható, vagy láthatatlan motívumok felkutatása a primer szekvenciában. profile, Pfam/Rfam, BLOCKS, CDD, COD. HMM.")
20
Szerkezeti információk megbízhatóbbá teszik a homológok azonosítását
Azonos funkcióhoz jobbára hasonló szerkezet járul Először csak azt vegyük figyelembembe, hogy nem minden egymást követő aminosav homológiája egyformán fontos a szerkezet és funkció szempontjából. Szerkezeti információk megbízhatóbbá teszik a homológok azonosítását Melyik fontos?

21
Hogy tudhatjuk meg, hogy melyek a fontos aminosavak?
A pontozásnál ne (ne nagyon) vegyük figyelembe a biológiai funkcióban részt nem vevő aminosavakat, és a konzerváltságnak megfelelő mértékben pontozzuk vagy súlyozzuk a lényegeseket! DNS-kötő fehérjék AT-hook motívuma Hogy tudhatjuk meg, hogy melyek a fontos aminosavak?
 vegyük figyelembe a biológiai funkcióban részt nem vevő aminosavakat, és a konzerváltságnak megfelelő mértékben pontozzuk vagy súlyozzuk a lényegeseket! DNS-kötő fehérjék AT-hook motívuma. Hogy tudhatjuk meg, hogy melyek a fontos aminosavak")
22
Multiple Alignment ! Egy fehérje: túl szemérmes.
Néhány: súg egy keveset. Hogyha mindet megkérdezed, hangos lesz a felelet. Multiple Alignment !

23
Multiple Alignment Heurisztikus módszereket alkalmazunk
Szimultán módszerek (m szekvencia összehasonlítása m dimenziós mátrixban) rendkívül időigényesek lennének: O (nm) Heurisztikus módszereket alkalmazunk
 rendkívül időigényesek lennének: O (nm) Heurisztikus módszereket alkalmazunk.")
24
Heurisztikus többszörös rendezők (Multiple alignment)
ClustalW, clustalv, clustalx (PC) (Thompson, Higgins, Gibson 1994) A szekvenciákból páronként távolságokat számít A távolságok alapján filogenetikai törzsfát (vezérfát) készít. A vezérfa szerinti távolságok alapján állapítja meg a többszörös rendezés sorrendjét A közeli szekvenciákat kisebb súllyal veszi figyelembe A BLOSUM mátrixok közül a távolságok alapján választ Oldallánc- és pozícióspecifikus pontozás MultAlin: (Corpet, 1988) Rekurzív eljárás: a kapott eredménybõl újraszámolja a vezérfát, ezzel új rendezést végez ezt addig ismétli, amíg a pontszám javul
 (Thompson, Higgins, Gibson 1994) A szekvenciákból páronként távolságokat számít A távolságok alapján filogenetikai törzsfát (vezérfát) készít. A vezérfa szerinti távolságok alapján állapítja meg a többszörös rendezés sorrendjét. A közeli szekvenciákat kisebb súllyal veszi figyelembe. A BLOSUM mátrixok közül a távolságok alapján választ. Oldallánc- és pozícióspecifikus pontozás. MultAlin: (Corpet, 1988) Rekurzív eljárás: a kapott eredménybõl újraszámolja a vezérfát, ezzel új rendezést végez. ezt addig ismétli, amíg a pontszám javul.")
26
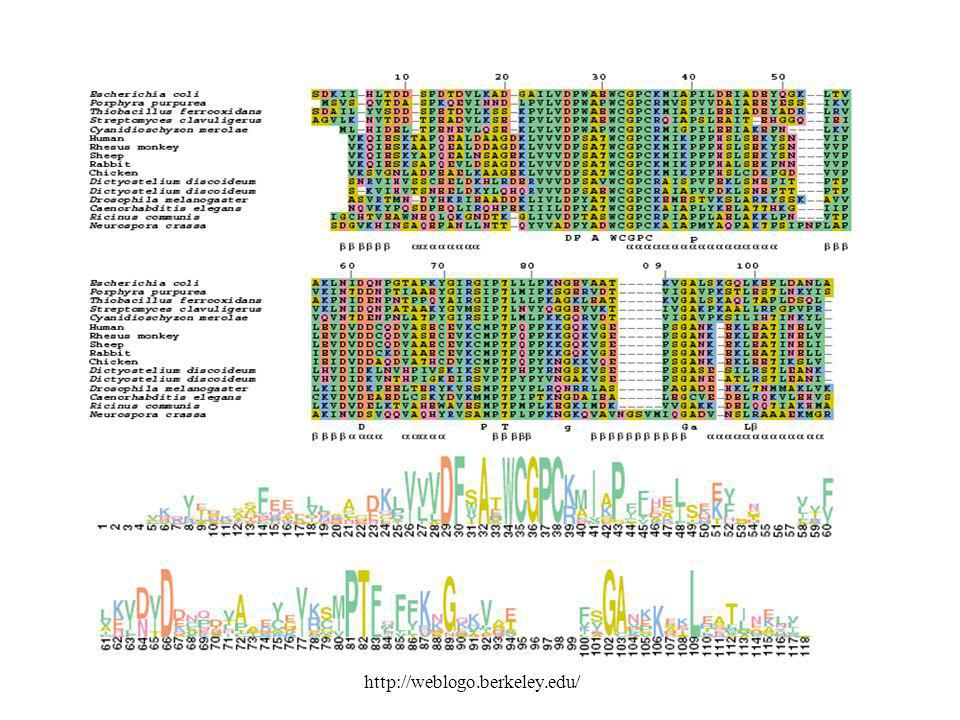
Multiple Alignment ! E. coli tioredoxin

28
Multiple Alignment ! Egy adott funkcióval kapcsolatba hozott állandó (?) aminosavak együttese: motif PROFILE pl ATP/GTP-bontó fehérjék foszfátkötő helye: P-loop Tioredoxin: WCGPC–[KR] + Kis adatbázis letölthető, tárolható; Egyszerű keresés (grep, regex) (FPAT, - nem hordoz elég információt Genbank mérete > 3x108 !! találat
 (FPAT, - nem hordoz elég információt. Genbank mérete > 3x108 !! 8000 találat.")
29
PSSM: Position Speific Scoring Matrix
Új homológok megtalálásához nem célszerű megkövetelni, hogy azok az ismert szekvenciákhoz tökéletesen hasonlítsanak: Valószínűségi módszereket kell alkalmaznunk, és Megfelelően nagy evolúciós távolságot átfogó, reprezentatív mintából származó aminosav-gyakoriságokat kell figyelembe venni PSSM: Position Speific Scoring Matrix Előfordulási valószínűség az adott pozícióban Előfordulási valószínűség az adott pozícióban × Subst.Mátrix (PAM/BLOSUM) Előfordulási valószínűség az adott pozícióban × Subst.Mátrix (PAM/BLOSUM) × AA gyakoriság Számos adatbázis elérhető: COD, CDD, BLOCKS, Pfam, Rfam, ...
 Előfordulási valószínűség az adott pozícióban × Subst.Mátrix (PAM/BLOSUM) × AA gyakoriság. Számos adatbázis elérhető: COD, CDD, BLOCKS, Pfam, Rfam, ...")
30
A profile-ok használata megkönnyíti:
Távoli homológok illesztését Az aktív helyek és a funkció meghatárzását Újabb homológok felkutatását A homológok osztályozását alcsoportokra Változékony aminosavak meghatározását (Ab) Térbeli szerkezetek meghatározását Kár, hogy a mátrixot a keresés előtt meg kell adni Rekurzió
 Térbeli szerkezetek meghatározását. Kár, hogy a mátrixot a keresés előtt meg kell adni Rekurzió.")
31
Dinamikusan változtatott, menet közben automatikusan származtatott scoring mátrix használata: PSI-Blast (Position-Specifc Iterated BLAST) Gapped BLAST az adatbázisban, egymástól függetlenül „Multiple Alignment” táblázat „Profile” előállítása ez utóbbiból Újra vizsgálja az adatbázist a Profile-lal Megtartja a szignifikáns találatokat Vissza a 2-es ponthoz, míg van változás, vagy a maximáils ciklusszámig
 Gapped BLAST az adatbázisban, egymástól függetlenül. „Multiple Alignment táblázat. „Profile előállítása ez utóbbiból. Újra vizsgálja az adatbázist a Profile-lal. Megtartja a szignifikáns találatokat. Vissza a 2-es ponthoz, míg van változás, vagy a maximáils ciklusszámig.")
32
HMM További BLAST-rokon programok:
blastpgp – protein profile előállítása formatrpsdb – profile adatbázis előállítása PHI-BLAST – Pattern-Hit-Initiated BLAST rpsblast, impala – reverse position-specific BLAST Keresés PSSM adatbázisban CDD Ungapped találatok kiterjesztése PSI-BLAST-nál kevésbé hatékony, de a BLAST-nál 100x gyorsabb lehet blastclust – automatikus szekvencia-”klaszterezés” bl2seq – két szekvencia között az optimális lokális illesztés A módszer, ami a BLAST-nál érzékenyebben talál rokonságot távoli homológok között tisztán szekvencia-adatok alapján: HMM

33
E. coli tioredoxin

34
Hidden Markov Model A pozícó-specifikus mátrixok használatának továbbfejlesztése egy matematikai eljárás képében, ahol minden pozícióban külön valószínűsége van az egyes aminosavaknak, inszerciónak és deléciónak A matematkai módszer alkalmazható szekvencia-illesztésre, homológia-keresésre, gén-keresésre, … ... beszéd- ill. írás felismerésre, rádiózavarszűrésre, stb.

35
A Hidden Markov Model általános szerkezete
A modellt fázisok, átmenetek és valószínűségek alkotják Minden fázist sorban meglátogatunk Az egyes fázisok egy-egy jelet bocsátanak ki Minden átmenetnek és kibocsátott jelnek meghatározott valószínűsége van; Σpi=1 A kibocsátott jelek láthatóak, míg a meglátogatott fázisok sorrendje rejtett A felhasznált lépések valószínűségének szorzata adja annak a valószínűségét, hogy a modell a kibocsátott jelek megfigyelhető sorrendjét szolgáltatja A valószínűségek az egyes fázisokban a többi fázistól függetlenek (távoli hatásokat nem vesz figyelembe)
")
36
Megfigyelt szekvencia
1. példa: “Egy (végtelen) szekvencia adott része AT-gazdag vagy GC-gazdag?” Megfigyelések: A DNS szekvenciának kétFÉLE szakasza van: AT-gazdag (1) és GC-gazdag (2) AT-gazdag régióhoz tartozó bázist jobbára hasonló fajta követ (és ford.) (ezért „szakasz”) AT-gazdag szakaszban is lehet G/C (és ford.) A GC-gazdag régiókban kicsit több a G, mint a C (ebben a kitalált esetben, ezen DNS-nek ezen a láncán) A GC-gazdag szakaszok rövidebbek, mint az AT-gazdag szakaszok Megfigyelt szekvencia Markov- lánc Ezek alapján három dolgot rendelünk egymáshoz: A megfigyelt szekvenciát A DNS szakasz „tulajdonságát” (1 v. 2) (AT v. GC) A modellt (Ezek közül kettőből TÖBB KÜLÖNBÖZŐ harmadik lenne származtatható) Modell
 szekvencia adott része AT-gazdag vagy GC-gazdag Megfigyelések: A DNS szekvenciának kétFÉLE szakasza van: AT-gazdag (1) és. GC-gazdag (2) AT-gazdag régióhoz tartozó bázist jobbára hasonló fajta követ (és ford.) (ezért „szakasz ) AT-gazdag szakaszban is lehet G/C (és ford.) A GC-gazdag régiókban kicsit több a G, mint a C (ebben a kitalált esetben, ezen DNS-nek ezen a láncán) A GC-gazdag szakaszok rövidebbek, mint az AT-gazdag szakaszok. Megfigyelt szekvencia. Markov- lánc. Ezek alapján három dolgot rendelünk egymáshoz: A megfigyelt szekvenciát. A DNS szakasz „tulajdonságát (1 v. 2) (AT v. GC) A modellt. (Ezek közül kettőből TÖBB KÜLÖNBÖZŐ harmadik lenne származtatható) Modell.")
37
Egy lehetséges „Markov-chain”
1. példa: “Egy (végtelen) szekvencia adott része AT-gazdag vagy GC-gazdag?” Két fázis Fázis-átmeneti valószínűségek A Modell Jelek és kibocsátási valószínűségeik Egy lehetséges „Markov-chain” Ezen valószínűségek szorzata adja meg annak a valószínűségét, hogy ez a HMM ezzel a fázis-sorrenddel ezt a szimbólum-szekvenciát generálja A megfigyelt szekvencia Kétféle szakasz: AT-gazdag (1) és GC-gazdag (2) Hasonló hasonlót követ … AT-gazdag szakaszban is lehet G/C (és ford.) A GC-gazdag régiókban kicsit több a G, mint a C A GC-gazdag szakaszok rövidebbek, mint az AT-gazdag szakaszok
 szekvencia adott része AT-gazdag vagy GC-gazdag Két fázis. Fázis-átmeneti valószínűségek. A Modell. Jelek és kibocsátási valószínűségeik. Egy lehetséges „Markov-chain Ezen valószínűségek szorzata adja meg annak a valószínűségét, hogy ez a HMM ezzel a fázis-sorrenddel ezt a szimbólum-szekvenciát generálja. A megfigyelt szekvencia. Kétféle szakasz: AT-gazdag (1) és GC-gazdag (2) Hasonló hasonlót követ … AT-gazdag szakaszban is lehet G/C (és ford.) A GC-gazdag régiókban kicsit több a G, mint a C. A GC-gazdag szakaszok rövidebbek, mint az AT-gazdag szakaszok.")
38
A megválaszolható kérdések
Az adott HMM milyen valószínűséggel generálja az adott szekvenciát? (Scoring) Melyik az optimális fázis szekvencia, amit a HMM használna az adott szekvencia generálásához? (Alignment) Egy nagy adathalmazt milyen paraméterekkel határozna meg legjobban az adott HMM? (Training) Két fázis Fázis-átmeneti valószínűségek Jelek és kibocsátási valószínűségeik A Modell Egy lehetséges „Markov-chain” A megfigyelt szekvencia
 Melyik az optimális fázis szekvencia, amit a HMM használna az adott szekvencia generálásához (Alignment) Egy nagy adathalmazt milyen paraméterekkel határozna meg legjobban az adott HMM (Training) Két fázis. Fázis-átmeneti valószínűségek. Jelek és kibocsátási valószínűségeik. A Modell. Egy lehetséges „Markov-chain A megfigyelt szekvencia.")
39
A kibocsátási és átmeneti valószínűségek meghatározása pl
A kibocsátási és átmeneti valószínűségek meghatározása pl. többszörös összerendezés (multiple alignment) alapján lehetséges
 alapján lehetséges.")
40
Az előző péda paraméterei
A DNS szekvenciának kétFÉLE szakasza van: AT-gazdag (1) és GC-gazdag (2) AT-gazdag régióhoz tartozó bázist jobbára hasonló fajta követ (és ford.) (ezért „szakasz”) AT-gazdag szakaszban is lehet G/C (és ford.) A GC-gazdag régiókban kicsit több a G, mint a C (ebben a kitalált esetben, ezen DNS-nek ezen a láncán) A GC-gazdag szakaszok rövidebbek, mint az AT-gazdag szakaszok Két fázis Fázis-átmeneti valószínűségek Jelek és kibocsátási valószínűségeik A Modell Egy lehetséges „Markov-chain” A megfigyelt szekvencia
 és. GC-gazdag (2) AT-gazdag régióhoz tartozó bázist jobbára hasonló fajta követ (és ford.) (ezért „szakasz ) AT-gazdag szakaszban is lehet G/C (és ford.) A GC-gazdag régiókban kicsit több a G, mint a C (ebben a kitalált esetben, ezen DNS-nek ezen a láncán) A GC-gazdag szakaszok rövidebbek, mint az AT-gazdag szakaszok. Két fázis. Fázis-átmeneti valószínűségek. Jelek és kibocsátási valószínűségeik. A Modell. Egy lehetséges „Markov-chain A megfigyelt szekvencia.")
41
2. példa: “5’ prime splice site”
Alignment Scoring

42
Minden lehetséges út kiszámítása rendkívül időigényes lenne, emiatt itt is speciális algoritmusokat alkalmazunk Scoring: Forward algoritmus A megelőző valószínűségek összege Alignment: Viterbi algoritmus A megelőző valószínűségek legnagyobbika + back-tracking Training Forward-Backward algoritmus Multiple alignment esetén lokális minimumok kivédésére további módszerek („noise injection”, „simulated annealing”)
")
43
Példa: Profile vs.HMM Vezérelv: Több adattal és alaposabb módszerrel pontosabb eredményt lehet kapni PROFILE HMM

44
3. példa: gén keresés

45
Néhány alapvető HMM (ungapped)
")
46
Főbb HMM-en alapuló programok és rokon web szolgáltatások

47
A HMM hiányossága Az átmeneti- és kibocsátási valószínűségek csak az aktuális fázis függvényei Emiatt távoli összefüggésekkel kapcsolatban nem használható RNS másodlagos szerkezet korrelált mutáció, pl. C-C További információt kell bevonni a távoli homológiák felderítesére: másodlagos, harmadlagos szerkezet

48
Az evolúció során fellépő mutációk a fehérje szerkezetében nem okoznak azonnal változásokat
Bizonyos szintű homológia mellett az adott funkciót hasonló szerkezet tartja (kivétel: konvergens evolúció!!!) Így a szerkezet felderítése segíthet a funkció megtalálásában
 Így a szerkezet felderítése segíthet a funkció megtalálásában.")
49
3D szerkezet Hasonló funkció Hasonló szerkezet
Hogyan határozhatjuk meg a szerkezetet a szekvenciából? G=H-TS 3D szerkezet a priori fizikai-kémiai módszerekkel (még) nem tudjuk meghatározni (kiszámolni) a fehérjék szerkezetét VIGYÁZAT!!! A fehérje szerkezetek nem nagyon stabilak Ált kJ/mol (5-15kcal/mol) stabilabbak, mint a denaturált foma (1-2 H2O-H2O H-H kötés)
 nem tudjuk meghatározni (kiszámolni) a fehérjék szerkezetét. VIGYÁZAT!!! A fehérje szerkezetek nem nagyon stabilak. Ált kJ/mol (5-15kcal/mol) stabilabbak, mint a denaturált foma (1-2 H2O-H2O H-H kötés)")
50
Közeli homológgal nem rendelkező fehérjék funkciójának meghatározása homológia alapján
~50507 protein szerkezet ismert (2008 dec. 11.) A hasonlóság elve alapján következtethetünk egyes sajátságokra szekvencia Fold recognition Másodlagos szerkezet SCOP, ... FASTA, BLAST PSI-BLAST, HMM CASP Vélt funkció KÍSÉRLET Mol.biol, Biochem
 A hasonlóság elve alapján következtethetünk egyes sajátságokra. szekvencia. Fold recognition. Másodlagos szerkezet. SCOP, ... FASTA, BLAST. PSI-BLAST, HMM. CASP. Vélt funkció. KÍSÉRLET. Mol.biol, Biochem.")
51
A fehérje molekula több doménből épülhet fel
A domének szerkezetileg és részben funkcionálisan független egységek Szerkezetük ill. funkciójuk külön-külön vizsgálandó Egy domén: gyakran több motif A domének független evoluciója következtében az egyes domének magasabb homológiát mutathatnak, mint a teljes fehérje A fehérjéket domének szerint csoportosíthatjuk (ld. később) Conserved Domain Database (CDD) (NCBI) ... Simple Modular Architecture Research Tool (SMART) ...
 Conserved Domain Database (CDD) (NCBI) ... Simple Modular Architecture Research Tool (SMART) ...")
52
Sasisekharan-Ramakrishnan-Ramchandran plot
Másodlagos szerkezeti elemek előrejelzése Sasisekharan-Ramakrishnan-Ramchandran plot A peptid kötés ált. sík (ált. trans ill a prolinnál cis) Az elvileg szabadon forgatható kötések energetikailag kedvező konformációi kijelölik kedvező másodlagos szerkezetet 6-20 αR konformáció: α hélix több β-konformáció: „extended szerkezet” – β strand 2 v. több β strand: β sheet
 Az elvileg szabadon forgatható kötések energetikailag kedvező konformációi kijelölik kedvező másodlagos szerkezetet αR konformáció: α hélix. több β-konformáció: „extended szerkezet – β strand 2 v. több β strand: β sheet.")
53
Másodlagos szerkezeti elemek előrejelzése
Hidrofil- és hidrofób oldalláncok váltakozása 2(β) ill. 3,5(α) aminosavanként α helix hidrofil- és hidrofób oldala: Helical wheel Hosszabb (15-30 aa) hidrofób régió: TM Az egyes aminosavaknak az egyes másodlagos szerkezetekben való eloszlási valószínűsége különbözik Sok egyéb Tusnady GE, Dosztanyi Z, Simon I. TMDET: web server for detecting transmembrane regions of proteins by using their 3D coordinates. Bioinformatics. 2005; 21(7):1276-7
 ill. 3,5(α) aminosavanként. α helix hidrofil- és hidrofób oldala: Helical wheel. Hosszabb (15-30 aa) hidrofób régió: TM. Az egyes aminosavaknak az egyes másodlagos szerkezetekben való eloszlási valószínűsége különbözik. Sok egyéb. Tusnady GE, Dosztanyi Z, Simon I. TMDET: web server for detecting transmembrane regions of proteins by using their 3D coordinates. Bioinformatics. 2005; 21(7):")
54
Másodlagos szerkezeti elemek előrejelzése
A kicsiny energetikai különbségek miatt pontosan nem lehet megjósolni (a határokat különösen) A megbízhatóság növelése érdekében minden lehetséges információt fel kell használni Új modellek, új algoritmusok (pl HMM) Homológ fehérjeszekvenciák (Multiple Alignment) Hasonló célú programok eredményei JPRED A módszerek értékelése CASP
 A megbízhatóság növelése érdekében minden lehetséges információt fel kell használni. Új modellek, új algoritmusok (pl HMM) Homológ fehérjeszekvenciák (Multiple Alignment) Hasonló célú programok eredményei. JPRED. A módszerek értékelése. CASP.")
55
A harmadlagos szerkezet meghatározására több független megközelítést alkalmaznak
(Ismert szerkezetű homológ esetén: homológia modellezés) 3D profiles (Adott szerkezetekben az egyes aminosavak környezete nem véletlen szerű. Az oldalláncok csoportosítása 6 csoportba, és a másodlagos szerkezetek 3 csoportba sorolása lehetővé teszi az aminosavak kódolását. Ezek között ezután „Folding Pattern” keresést lehet végezni) Threading (készítsünk szerkezeteket a kérdéses molekulából, majd „gap”-ek közbeiktatásával illesszük ezeket az ismert 3D szerkezetekre) ...
 3D profiles (Adott szerkezetekben az egyes aminosavak környezete nem véletlen szerű. Az oldalláncok csoportosítása 6 csoportba, és a másodlagos szerkezetek 3 csoportba sorolása lehetővé teszi az aminosavak kódolását. Ezek között ezután „Folding Pattern keresést lehet végezni) Threading (készítsünk szerkezeteket a kérdéses molekulából, majd „gap -ek közbeiktatásával illesszük ezeket az ismert 3D szerkezetekre) ...")
56
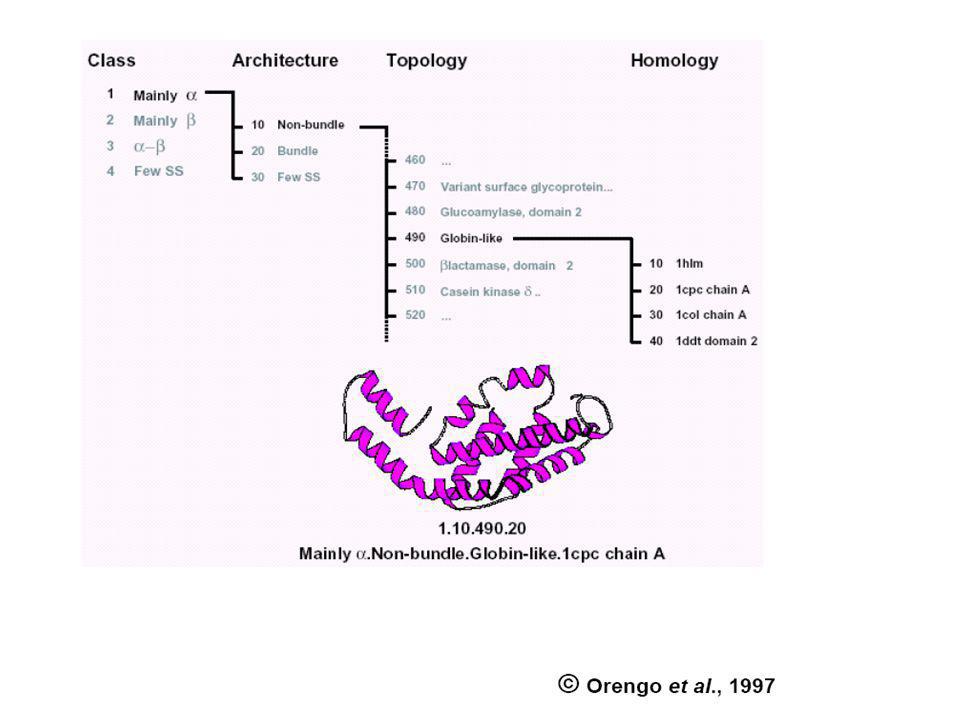
Harmadlagos szerkezetek osztályozása (egymásra kereszthivatkozó) speciális adatbázisokkal történik
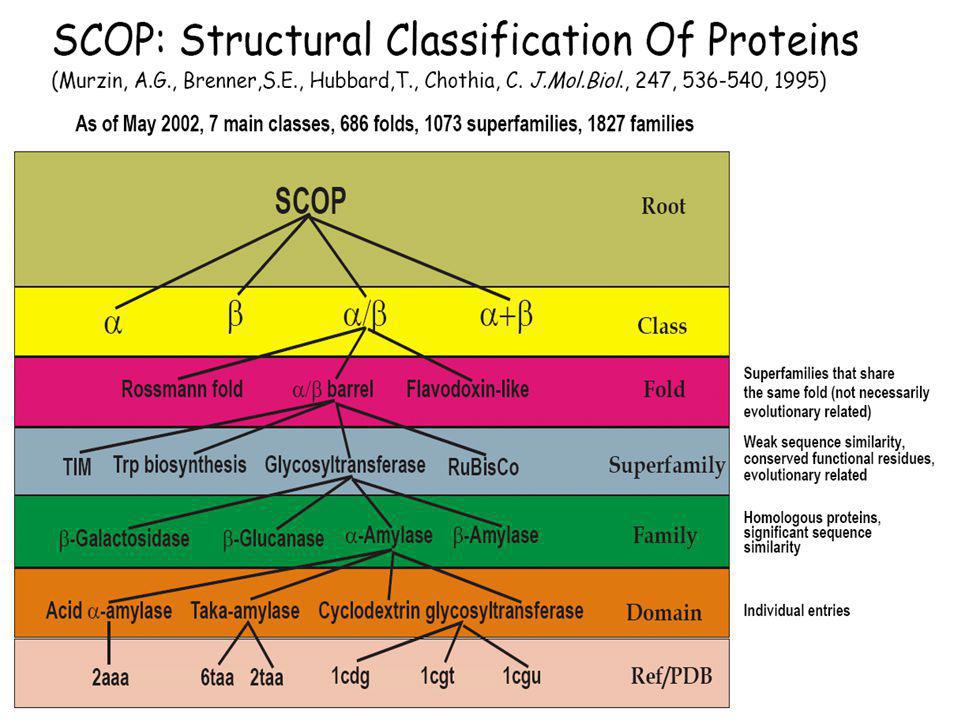
CATH: protein domain szerkezetek hierarchikus osztályozása négy szinten: Class(C), Architecture(A), Topology(T) and Homologous superfamily (H). SCOP: (Structural Classification of Proteins) Domains (a PDB adatbázisból) Families (Homológ domének. Szekveniájuk, szerkezetük ill. Funkciójuk hasonlósága közös őst valószínűsít) Superfamilies (Hasonló szerkezetű és funkciójú fehérjék családjai, ahol a rokonság valószínűsíthető, de nem bizonyított) Folds (hasonló topológiájú „Superfamilies”) CLASS (all-α; all-β; α/β; α+β, multi-domén; membrán- és sejtfelszín; egyéb kis proteinek, peptidek;) ...
, Architecture(A), Topology(T) and Homologous superfamily (H). SCOP: (Structural Classification of Proteins) Domains (a PDB adatbázisból) Families (Homológ domének. Szekveniájuk, szerkezetük ill. Funkciójuk hasonlósága közös őst valószínűsít) Superfamilies (Hasonló szerkezetű és funkciójú fehérjék családjai, ahol a rokonság valószínűsíthető, de nem bizonyított) Folds (hasonló topológiájú „Superfamilies ) CLASS (all-α; all-β; α/β; α+β, multi-domén; membrán- és sejtfelszín; egyéb kis proteinek, peptidek;) ...")
60
Protein: Flavodoxin from Anabaena
Lineage: Root: scop Class: Alpha and beta proteins (a/b) [51349] Mainly parallel beta sheets (beta-alpha-beta units) Fold: Flavodoxin-like [52171] 3 layers, a/b/a; parallel beta-sheet of 5 strand, order 21345 Superfamily: Flavoproteins [52218] Family: Flavodoxin-related [52219] binds FMN Protein: Flavodoxin [52220] Species: Anabaena, pcc 7119 and 7120 [52223] PDB Entry Domains: 1obo complexed with fmn, so4; mutant chain a [86776] chain b [86777] 1rcf [31170] complexed with fmn, so4 1dx9 apo form complexed with so4; mutant chain a [31171] chain b [31172] chain c [31173]
 [51349] Mainly parallel beta sheets (beta-alpha-beta units) Fold: Flavodoxin-like [52171] 3 layers, a/b/a; parallel beta-sheet of 5 strand, order Superfamily: Flavoproteins [52218] Family: Flavodoxin-related [52219] binds FMN. Protein: Flavodoxin [52220] Species: Anabaena, pcc 7119 and 7120 [52223] PDB Entry Domains: 1obo complexed with fmn, so4; mutant. chain a [86776] chain b [86777] 1rcf [31170] complexed with fmn, so4. 1dx9 apo form complexed with so4; mutant. chain a [31171] chain b [31172] chain c [31173]")
62
A bioinformatika is kísérletes tudomány
Kizárólag ab inito módszerekkel (energetikai minimalizálással) a teljes molekulák szerkezetének meghatározása nem megoldható „Knowledge-based” módszereket alkalmazunk A „knowledge” egyre bűvül az ezen alapuló módszerek is folyamatosan fejlődnek A módszereket tesztelni kell (in silico KÍSÉRLET) CASP: Critical Assessment of Techniques for Protein Structure Prediction 2 évente végzett „blind test” Különböző nehézségi kategóriákban meghirdetett szekvenciák Titokban tartott, újonnan meghatározott szerkezetekkel
 a teljes molekulák szerkezetének meghatározása nem megoldható. „Knowledge-based módszereket alkalmazunk. A „knowledge egyre bűvül az ezen alapuló módszerek is folyamatosan fejlődnek. A módszereket tesztelni kell (in silico KÍSÉRLET) CASP: Critical Assessment of Techniques for Protein Structure Prediction. 2 évente végzett „blind test Különböző nehézségi kategóriákban meghirdetett szekvenciák. Titokban tartott, újonnan meghatározott szerkezetekkel.")
64
példa: HMMSPECTR A CASP4 eredményekkel összehasonlítva igen jó teljesítményt mutat Két, hasonló funkciójú de nagyon különböző szekvenciájú fehérje hasonló szerkezeti elemeinek kimutatása:

65
A bioinformatikában igen gyakran
több különálló programnak sok szekvenciával, sokszori futtatása során keletkező rengeteg, jellemzően szöveges file-t kell értelmezni, értékelni, ezek alapján dönteni a továbi lépésekről. Ezt a tevékenységet gyakran célszerű (elkerülhetetlen) számítógépekre bízni PERL
 számítógépekre bízni. PERL.")


 DNS-ből,>")















 Bihari Péter.>")


 Csernetics Árpád Bioinformatika SZIT 2005. ápr. 18.>")