Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
Metabolic disorders

2
A metabolic disorder is a medical disorder which affects the production of energy within individual human (or animal) cells. Most metabolic disorders are genetic, though a few are "acquired" as a result of diet, toxins, infections, etc. Genetic metabolic disorders are also known as inborn errors of metabolism.

3
The largest classes of metabolic disorders are:
- Disorders of carbohydrate metabolism - Disorders of amino acid metabolism - Disorders of fatty acid oxidation and mitochondrial metabolism - Disorders of porphyrin metabolism - Disorders of purine or pyrimidine metabolism - Disorders of steroid metabolism - Disorders of mitochondrial function - Disorders of peroxisomal function - Lysosomal storage disorders

4
Disorders of carbohydrate metabolism

5
Pyruvate metabolism and gluconeogenesis
Glucose-P-isomerase deficiency Anaemia, neurologic symptoms (musc. spasticity) -Pyruvate kinase deficiency hemolytic anaemia Pyruvate Dehydrogenase Deficiency (PDHA) It affects a gene which codes for a critical enzyme complex, the Pyruvate dehydrogenase complex (PDC) PDHA causes Lactic acidosis; large amounts of lactic acid in the blood but with a normal pyruvate/lactate ratio. Symptoms are varied, and include developmental defects (especially of the brain and nervous system), muscular spasticity and early death.
 -Pyruvate kinase deficiency. hemolytic anaemia. Pyruvate Dehydrogenase Deficiency (PDHA) It affects a gene which codes for a critical enzyme complex, the Pyruvate dehydrogenase complex (PDC) PDHA causes Lactic acidosis; large amounts of lactic acid in the blood but with a normal pyruvate/lactate ratio. Symptoms are varied, and include developmental defects (especially of the brain and nervous system), muscular spasticity and early death.")
6
Pyruvate carboxylase deficiency
is an inherited disorder that causes lactic acid and other potentially toxic compounds to accumulate in the blood. High levels of these substances can damage the body's organs and tissues, particularly in the nervous system. Type A: Characteristic features include developmental delay and a buildup of lactic acid in the blood (lactic acidosis). Increased acidity in the blood can lead to vomiting, abdominal pain, extreme tiredness (fatigue), muscle weakness, and difficulty breathing. Children with pyruvate carboxylase deficiency type A typically survive only into early childhood. Type B Pyruvate carboxylase deficiency type B has life-threatening signs and symptoms that become apparent shortly after birth. This form of the condition has been reported mostly in Europe, particularly France. Affected infants have severe lactic acidosis, a buildup of ammonia in the blood (hyperammonemia), and liver failure. They experience neurological problems including weak muscle tone (hypotonia), abnormal movements, seizures, and coma. Infants with this form of the condition usually survive for less than 3 months after birth.
. Increased acidity in the blood can lead to vomiting, abdominal pain, extreme tiredness (fatigue), muscle weakness, and difficulty breathing. Children with pyruvate carboxylase deficiency type A typically survive only into early childhood. Type B Pyruvate carboxylase deficiency type B has life-threatening signs and symptoms that become apparent shortly after birth. This form of the condition has been reported mostly in Europe, particularly France. Affected infants have severe lactic acidosis, a buildup of ammonia in the blood (hyperammonemia), and liver failure. They experience neurological problems including weak muscle tone (hypotonia), abnormal movements, seizures, and coma. Infants with this form of the condition usually survive for less than 3 months after birth.")
7
Drug induced hemolytic anemia

8
dehydrogenase deficiency
Glucose 6-phosphate dehydrogenase deficiency -inherited disease characterized by hemolytic anemia due to inability to detoxify oxidizing agents -most common disease-producing enzyme abnormality in humans (>200 million people worldwide, ~7%; ~2% of U.S. population) -X-linked deficiency caused by >300 different mutations in the G6PD gene -only some mutations cause clinical disease -life span of individuals with G6PD deficiency shortened somewhat due to complications of chronic hemolysis -G6PD deficiency has been maintained in the human gene pool by the evolutionary advantage of increased resistance to falciparum malaria in female carriers of the mutations in the tropics -geographic distribution (highest in tropical Africa, Asia, Middle East, Mediterranean and Papua New Guinea) follows sickle cell trait (also confers relative resistance to malaria)
 -X-linked deficiency caused by >300 different mutations in the G6PD gene. -only some mutations cause clinical disease. -life span of individuals with G6PD deficiency shortened somewhat due to complications of chronic hemolysis. -G6PD deficiency has been maintained in the human gene pool by the evolutionary advantage of increased resistance to falciparum malaria in female carriers of the mutations in the tropics. -geographic distribution (highest in tropical Africa, Asia, Middle East, Mediterranean and Papua New Guinea) follows sickle cell trait (also confers relative resistance to malaria)")
9
Bodies in Red Blood Cells
Formation of Heinz Bodies in Red Blood Cells - RBCs generate superoxide and other ROS during non-enzymatic oxidation of hemoglobin (Hb) to metHb by spontaneous transfer of an electron from the Fe2+ in Hb to bound O2 - RBCs depend on G6PD for generating NADPH to re-reduce glutathione to protect against this oxidative stress - G6PD deficiency results in disulfide linked aggregates of Hb forming on the red cell membrane - mechanical stress from lack of deformability in small capillaries and ROS peroxidation of membrane lipids
 to metHb by spontaneous transfer of an electron from the Fe2+ in Hb to bound O2. - RBCs depend on G6PD for generating. NADPH to re-reduce glutathione to. protect against this oxidative stress. - G6PD deficiency results in disulfide linked aggregates of Hb forming on. the red cell membrane. - mechanical stress from lack of. deformability in small capillaries and. ROS peroxidation of membrane lipids.")
10
Precipitating factors for G6PD
deficiency disease Oxidant Drugs (A3): AAA= Antibiotics (e.g. sulfamethoxazole) , Antimalarials (e.g. primaquine), Antipyretics (acetanilid, not aspirin or acetominophen) Favism: Mediterranean variant is susceptible to hemolytic affects of ingesting fava bean purine glycosides (vicine and isouramil) which react with reduced glutathione, decreasing GSH levels Infection: inflammatory response to infection results in the generation of free radicals (ROS) by macrophages and neutrophils; ROS can diffuse into RBCs and induce oxidative damage Neonatal jaundice: results from impaired hepatic catabolism or increased production of bilirubin (heme degradation product from hemoglobin, myoglobin, cytochromes)
: AAA= Antibiotics (e.g. sulfamethoxazole) , Antimalarials (e.g. primaquine), Antipyretics (acetanilid, not aspirin or acetominophen) Favism: Mediterranean variant is susceptible to hemolytic affects of ingesting fava bean purine glycosides (vicine and isouramil) which react with reduced glutathione, decreasing GSH levels. Infection: inflammatory response to infection results in the generation of free radicals (ROS) by macrophages and neutrophils; ROS can diffuse into RBCs and induce oxidative damage. Neonatal jaundice: results from impaired hepatic catabolism or increased production of bilirubin (heme degradation product from hemoglobin, myoglobin, cytochromes)")
11
Classification of Glucose-6-phosphate dehydrogenase deficiency

12
Common Hexoses Sucrose (table sugar): glucose + fructose and
Lactose: (dairy products) glucose + galactose
 glucose + galactose.")
13
Sucrose (Table Sugar) α-D-glucopyranosyl-(1→2)-β-D-fructofuranoside
 α-D-glucopyranosyl-(1→2)-β-D-fructofuranoside")
14
Cleavage of Sucrose (α-glucosidase or invertase)
")
15
Muscle Metabolism of Fructose (Anaerobic Glycolysis) Muscle Hexokinase can accept fructose as a substrate
 Muscle Hexokinase can accept fructose as a substrate")
16
Liver Metabolism of Fructose I Glucokinase, the liver isoform of hexokinase cannot transform fructose

17
Liver Metabolism of Fructose II

18
Fructose Intolerance Too Much Fructose
Fructose-1-P Aldolase ( Aldolase B) is rate-limiting Depletion of Pi Reduction in [ATP] Increase in glycolysis Accumulation of lactate (acid) in blood Fructose-1-P Aldolase Deficiency (Genetic Disease) + Liver damage
 is rate-limiting. Depletion of Pi. Reduction in [ATP] Increase in glycolysis. Accumulation of lactate (acid) in blood. Fructose-1-P Aldolase Deficiency (Genetic Disease) + Liver damage.")
19
Galactose Metabolism

20
Lactose Metabolism (Dairy Products)
Glycolytic Enzymes are specific and do not recognize galactose!

21
Need Epimerization

22
Phosphorylation of Galactose
2 A T P D 3 = G a l c t o k i n s e - 1

23
Activation of Galactose

24
Epimerization of UDP-Galactose

25
Lactose intolerance (or hypolactasia) is the term used to describe a decline in the level of lactase, an enzyme needed for proper metabolization of lactose (a sugar that is a constituent of milk and other dairy products), in human beings. Lactose intolerance in varying degrees is physiologically normal in adult mammals, including many human beings. However, certain ethnic groups - particularly those of European descent - continue to produce lactase throughout their lives. Without lactase, the lactose in milk and dairy remains uncleaved and unabsorbed. Lactose cannot pass easily through the intestinal wall into the bloodstream, so it remains in the intestines. Soon, enteral bacteria adapt to the relative abundance of lactose (relative to other usable sugars like glucose) and their operons quickly switch over to lactose metabolism. Along the way they produce copious amounts of gas by fermentation. The gas causes a range of unpleasant abdominal symptoms, including stomach cramps, bloating, flatulence and diarrhea. Like other unabsorbed sugars, e.g. mannitol, the lactose raises the osmotic pressure of the colon contents, preventing the colon from reabsorbing water and hence causing a laxative effect to add to the excessive gas production.
 and their operons quickly switch over to lactose metabolism. Along the way they produce copious amounts of gas by fermentation. The gas causes a range of unpleasant abdominal symptoms, including stomach cramps, bloating, flatulence and diarrhea. Like other unabsorbed sugars, e.g. mannitol, the lactose raises the osmotic pressure of the colon contents, preventing the colon from reabsorbing water and hence causing a laxative effect to add to the excessive gas production.")
26
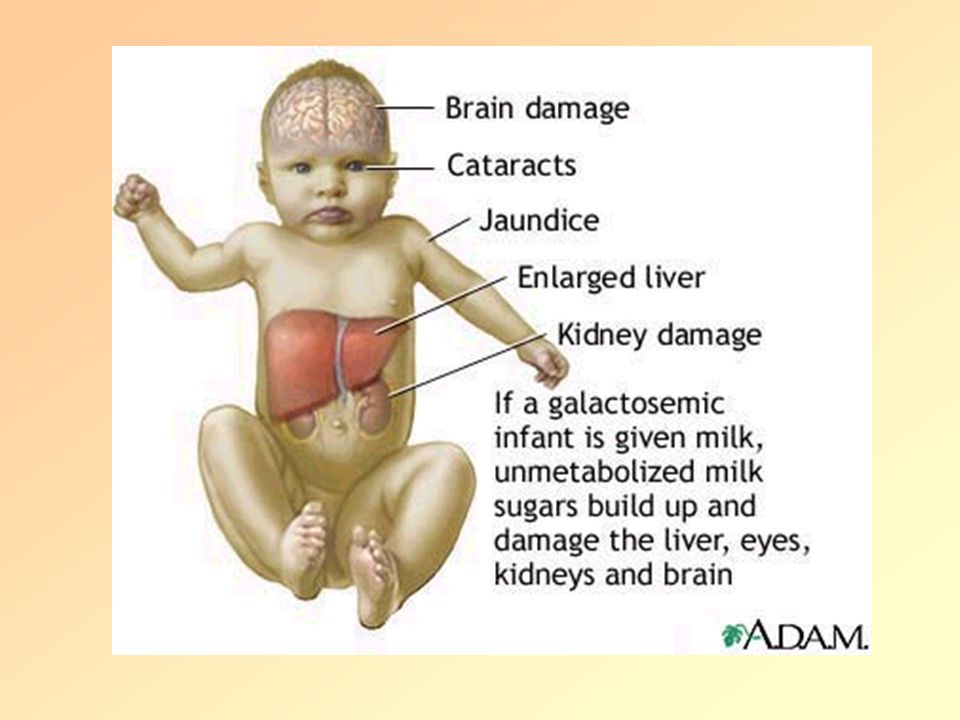
Galactosemia (Mental Retardation and Death)
Treatment Galactose-free diet (reversal of all symptoms except mental retardation)
")
28
Cataracts

29
Glycogen storage disease (synonyms: glycogenosis, dextrinosis) is any one of several inborn errors of metabolism that result from enzyme defects that affect the processing of glycogen synthesis or breakdown within muscles, liver, and other cell types. Types There are nine diseases that are commonly considered to be glycogen storage diseases. (Although glycogen synthase deficiency does not result in storage of extra glycogen in the liver, it is often classified with the GSDs as type 0 because it is another defect of glycogen storage and can cause similar problems.)
")
30
Glycogen Storage Diseases
Type 0 Type IV Type I Type VII Type II

32
Glycogen Storage 1: von Gierke disease
Accumulation of glycogen in liver and kidney => hepatomegaly Hypoglycemia Enzyme deficiency: Glucose 6 phosphatase Accumulation: Glycogen

33
Infantile onset < 12 months
Pompe Disease Signs & Symptoms Infantile onset < 12 months Late onset > 12 months Daytime somnolence Morning headache Head lag Shortness of breath/ sleep apnea Respiratory insufficiency Enlarged tongue Cardiomegaly/ cardiomyopathy Scapular winging Respiratory insufficiency Scoliosis Delayed motor development Low back pain Gait abnormality Organomegaly Muscle weakness Muscle weakness Hirschhorn R, Reuser AJJ. In: The Metabolic and Molecular Bases of Inherited Disease. 2001:

34
Glycogen storage 2: Pompe disease
Muscle hypotonia Splenomegaly cardiomegaly Death before age 3 of cardiorespiratory failure Intractable Hypoglycemia Enzyme deficiency: alpha-1,4 glucosidase Accumulation: Glycogen

35
Pompe Disease Enlarged tongue/ lax facial features
Signs & Symptoms Data on file, Genzyme Corporation. Enlarged tongue/ lax facial features Courtesy of R. R. Howell, MD. Hypotonia/head lag/ floppy baby Cardiomegaly

36
Pompe Disease Lordosis / Scoliosis Weak pelvic girdle muscles
Signs & Symptoms Lordosis / Scoliosis Weak pelvic girdle muscles

37
Inheritance Pompe Disease is autosomal recessive: Father Carrier
Mother Carrier 1 2 3 4 Affected Individual (25%) Unaffected Carriers (50%) Unaffected Noncarrier (25%) Unaffected (75%)
 Unaffected. Carriers. (50%) Unaffected. Noncarrier. (25%) Unaffected. (75%)")
38
Glycogen Storage 3: Cori Disease
Glycogen accum in heart Glycogen in liver -> Hepatomegaly hypoglycemia Glycogen in skeletal muscle Stunted growth Enzyme deficiency: Amylo-1,6-glucosidase Accumulation: Glycogen

39
Case Description A female baby was delivered normally after an uncomplicated pregnancy. At the time of the infant’s second immunization, she became fussy and was seen by a pediatrician, where examination revealed an enlarged liver. The baby was referred to a gastroenterologist and later diagnosed to have Glycogen Storage Disease Type IIIB

40
Glycogen Storage Disease Type IIIb
Deficiency of debranching enzyme in the liver needed to completely break down glycogen to glucose Hepatomegaly and hepatic symptoms Usually subside with age Hypoglycemia, hyperlipidemia, and elevated liver transaminases occur in children

41
Glycogen Storage 4: McArdle Syndrome
Enzyme deficiency: Muscle phosphorylase Accumulation: Glycogen Muscle cramps and weakness after exercise

42
Disorders of amino acid metabolism

43
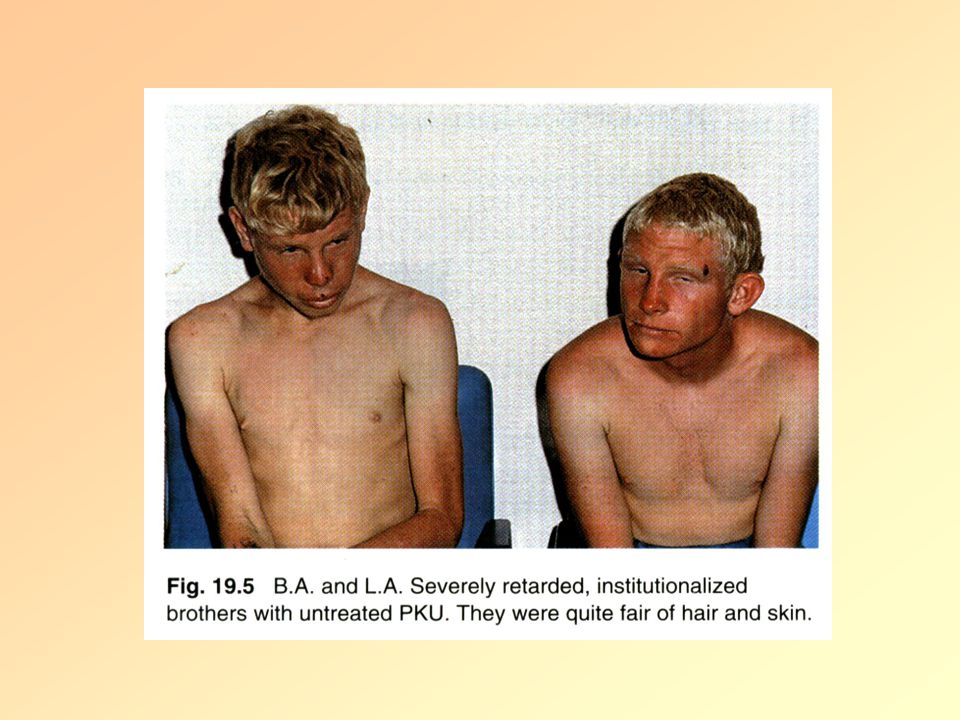
Phenylketonuria (PKU)
Phenylalanin hydroxylase (PAH) defected 12. chromosome, >400 diff. mutations (autosomal recessive disorder) Symptoms: Without treatment severe mental retardation (very low IQ) epilepsy hypopoigmentation: blond, blue eyes Eczema
 defected. 12. chromosome, >400 diff. mutations. (autosomal recessive disorder) Symptoms: Without treatment severe mental retardation (very low IQ) epilepsy. hypopoigmentation: blond, blue eyes. Eczema.")
44
Types of PKU 1./ Classical PKU : Phe level in the blood > 10mg%- life-long treatment 2./ Non PKU hyperfenylalaninaemia: Blood Phe 2-10mg %: not so strict diet 3./ Atypical PKU, PAH-cofactor tetrahydrobiopterin, (BH4) def: retardation during pregnancy, early symptoms, fever, spasticity, mental and developmental retardation. Th: BH4 , diet not enough, serotonin, dopamin supplementation
 def: retardation during pregnancy, early symptoms, fever, spasticity, mental and developmental retardation. Th: BH4 , diet not enough, serotonin, dopamin supplementation.")
45
Without phenylalanine hydroxylase PHE→Tyr not occur Tyr deficiency may lead to hypopigmentation
High Phe can cause neurologic damage

48
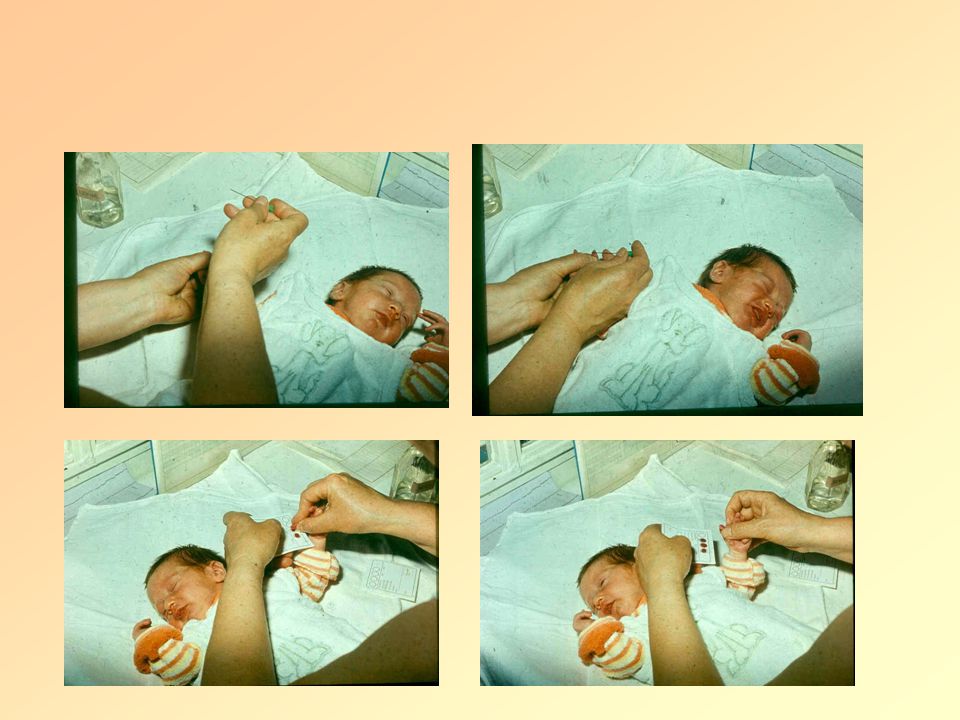
Newborn screening Important- early diagnosis and treatment of affected infants resuling in normal growth and development! Since 1965! At 5. day or after feeding Method: GUTHRIE test: Bacillus Subtilis grows only Phe containing broth.

49
Guthrie test

51
Urine test (Ferri III-chloride)
")
52
Diet in PKU Protein: 60 – 80 % - low protein content
- fruits, vegetables Phe level: 4 – 8 mg %. NO: milk – diary products, meat, fish, egg, bean, chocolate starch (potato, kukorica) Vitamins, nyomelem Chalories: ~ 35 % fat, ~ 15 % tápszer0 50 % CHO Anyatej: csak lefejve, ml/nap, ellenőrizve phenylalanin szintet Szoptatás= ellenőrizetlen mennyiség bevitel
 Vitamins, nyomelem. Chalories: ~ 35 % fat, ~ 15 % tápszer0 50 % CHO. Anyatej: csak lefejve, ml/nap, ellenőrizve phenylalanin szintet. Szoptatás= ellenőrizetlen mennyiség bevitel.")
53
PKU gyermek, diétán egészséges
Fenilalanin szegény étrend időtartama: korábban éves korig, ma tudjuk, egész életre szólóan ajánlott KÖTELEZŐ: PKU-s felnőtt egykori gondozott a várandósság Teljes ideje alatt: az esetleg egészséges magzatot is károsítja az anyai magasabb fenilalanin!!

54
Albinism

55
Albinism Formation of little or no skin pigment
Classical defect is tyrosinase (tyrosine hydroxylase) Autosomal recessive 1: 20000
 Autosomal recessive. 1:")
56
Albinism Light hair and skin UV light (esp. 280-320 nm)
Increased light sensitivity Increased risk for skin cancer Normal growth and development

58
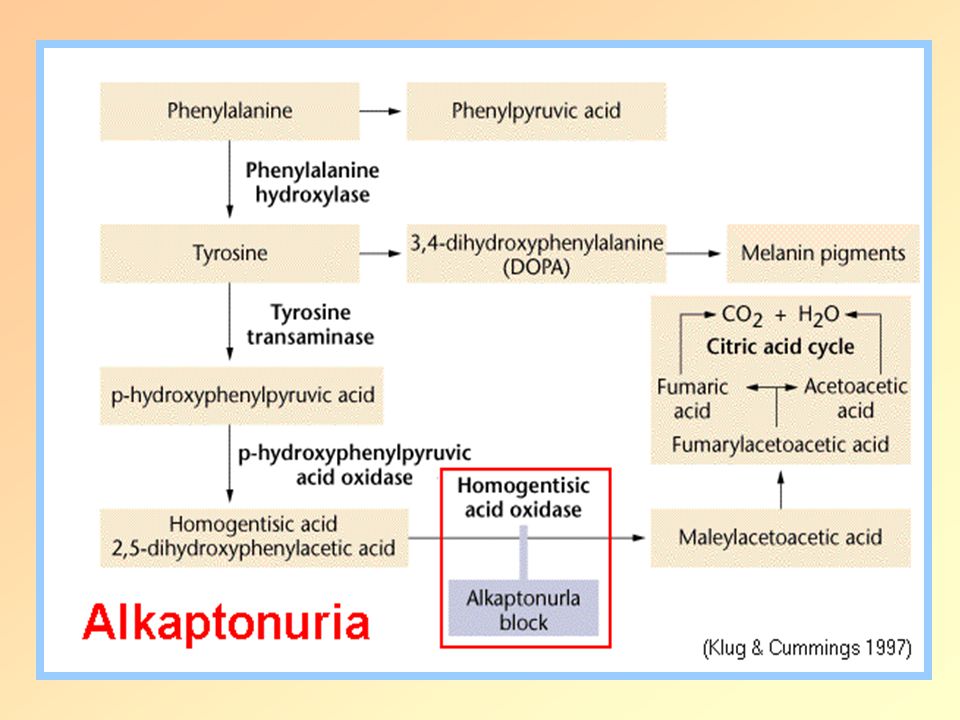
Alkaptonuria Autosomal recessive Lack of homogentisate oxidase
Increased homogentisic acid

59
Alcaptonuria Symptoms:
Homogentisate appears in the urine deposited in cartilage- inflammation in arth Black dots on cornea Kidney Polymerized homogentisate on the skin=Ochronosis Ocular ochronosis in alkaptonuric patient Polymerized homogentisate in ear cartilage

60
Typical alkaptonuric position

61
MAPLE SYRUP URINE DISEASE
Most common BCAA disorder (1/185,000) Defective branched-chain ketoacid dehydrogenase Similar to PDH with 3 enzyme activities Thiamine deficiency can produce same result Keto acids that accumulate smell like burn maple syrup BCAA also accumulate Mental retardation Untreated leads to death
 Defective branched-chain ketoacid dehydrogenase. Similar to PDH with 3 enzyme activities. Thiamine deficiency can produce same result. Keto acids that accumulate smell like burn maple syrup. BCAA also accumulate. Mental retardation. Untreated leads to death.")
62
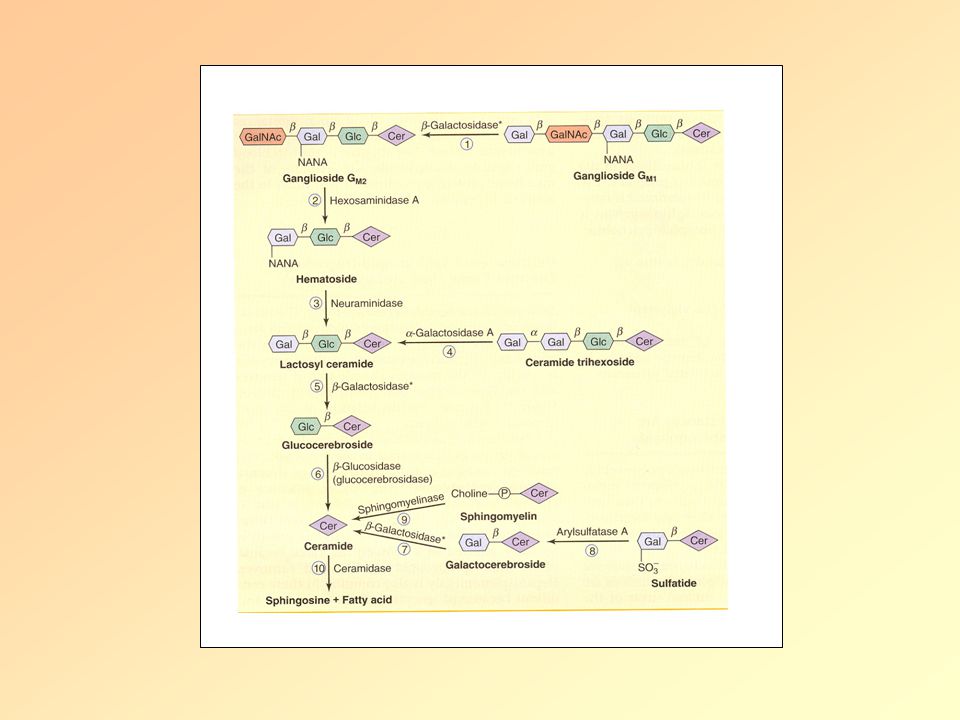
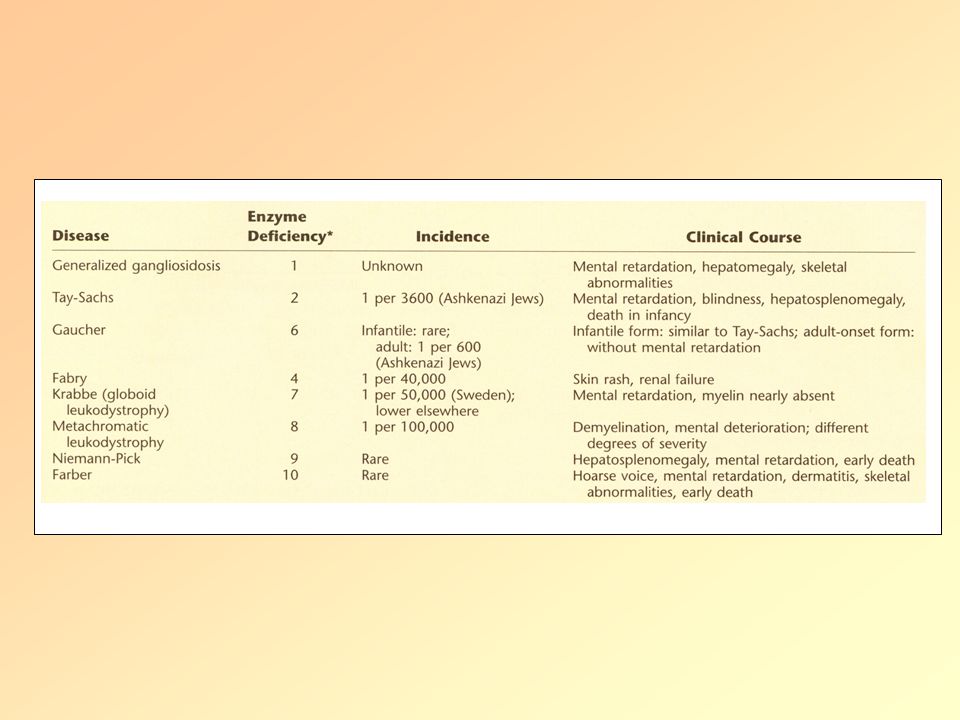
Lipid anyagcsere betegségek Lipid/Lizoszómális tárolási betegségek
Definicíó: öröklött anyagcsere betegség, amelyben a lipidek bizonyos szövetekben, sejtekben kóros mértékben felszaporodnak. A betegeknek vagy nincs egy bizonyos enzimből elegendő mennyiség, vagy olyan enzimet termelnek, ami nem működik megfelelően. Hosszú idő alatt a nagy mennyiségben tárolt lipidek károsodásokat okoznak, főleg az agyban, perifériás ideg rendszerben, májban, lépben, csontvelőben. Lipidek: Zsírszerű anyagok, amelyek fontosak a sejtmembránok felépítésében és a myelint burkolják, így védik az idegeket. A sejtek lizoszómákban tárolják őket, és ha szükséges energia forrásként használjuk őket. Ezért lizoszómális tárolási betegségnek is nevezzük őket. A lipid tárolási zavarokon kívül léteznek még mucolipoidosisok (nagy mennyiségű cukor és lipid halmozódik fel), valamint mucopolysaccharidosisok (cukor halmozódik fel).
, valamint mucopolysaccharidosisok (cukor halmozódik fel).")
65
Lipid tárolási betegségek II.
Tipusai: Szfingolipid tárolási bet./Gangliozidozisok: -GM1 gangliozidozis -GM2 gangliozidozis: -Tay-Sachs -Sandhoff Gaucher Niemann-Pick Fabry Farber Krabbé Szfingolipid

66
GM1 gangliozidozis β-galaktozidáz hiánya: β-galaktozidból→ monoszacharid A perifériás, de főleg a központi idegrendszer sejtjei abnormális mennyiségben raktároznak lipideket. Szubsztrátjai közé tartozik: GM1 gangliozid, laktóz, különböző glikoproteinek. Megjelenési formái és tünetei: Korai infantilis: süketség, vakság, tüdőgyulladás Késői infantilis: dementia, beszéd zavar Felnőttkori: dystonia

67
GM2 gangliozidozis:Tay-Sachs, Sandhoff
Β-hexózaminidáz-A hiány: gangliozidokat bont, a lizoszómákban (főleg neuronok) található A GM2 gangliozid lebontásához 3 fehérje kell, ebből 2 a hexóaminidáz alegysége a 3. kofaktora az enzimnek: GM2 aktivátor protein (glikolipid transzport fehérje). Ha a 3 fehérjéből valamelyik nem működik vagy nincs elegendő mennyiségben→tárolási betegség. Tay-Sachs: ált. az enzim foldingja nem megfelelő, így nincs intracelluláris transzport Megjelenési formák és tünetek: Infantilis: süketség, vakság, nyelési képtelenség, atrófia Serdülőkori: dysarthria, dysphagia, spasticitás, ataxia Felnőttkori: schizofrénia, pszichózis, bénulás Szűrés vérből!
 található. A GM2 gangliozid lebontásához 3 fehérje kell, ebből 2 a hexóaminidáz alegysége. a 3. kofaktora az enzimnek: GM2 aktivátor protein (glikolipid transzport fehérje). Ha a 3 fehérjéből valamelyik nem működik vagy nincs elegendő mennyiségben→tárolási betegség. Tay-Sachs: ált. az enzim foldingja nem megfelelő, így nincs intracelluláris transzport. Megjelenési formák és tünetek: Infantilis: süketség, vakság, nyelési képtelenség, atrófia. Serdülőkori: dysarthria, dysphagia, spasticitás, ataxia. Felnőttkori: schizofrénia, pszichózis, bénulás. Szűrés vérből!")
68
GM2 gangliozidozis II. Sandhoff-betegség:
β-Hexózaminidáz-B hiány: agy és gerincvelői neuronokat progresszíven pusztítja Tünetek: Mentális retardáció, bénulás, cseresznye piros folt a retinán, (organomegalia)
")
69
Gaucher-betegség I. A lizoszómális glucocerebrosidase(=β-glucosidáz) hiánya Glukocerebrozid(=glucosylceramide) lebontása zavart (vörös és fehérvérsejtek membránjának alkotója) A macrophagok nem tudják ezeket a sejteket teljesen lebontani→glucocerebrozid felhalmozódik→Gaucher-sejt: fénymikroszkóp alatt „felgyűrt-papír”-ra emlékeztetnek Az idegrendszerben szintén felhalmozódik a glucocerebrozid a lipidekkel együtt Parkinson-kór, non-Hodgkin-lymphoma, Melanoma, pancreas tumor gyakoribb Glucocerebrosidase
 lebontása zavart (vörös és fehérvérsejtek. membránjának alkotója) A macrophagok nem tudják ezeket a sejteket teljesen lebontani→glucocerebrozid felhalmozódik→Gaucher-sejt: fénymikroszkóp alatt „felgyűrt-papír -ra emlékeztetnek. Az idegrendszerben szintén felhalmozódik a glucocerebrozid a lipidekkel együtt. Parkinson-kór, non-Hodgkin-lymphoma, Melanoma, pancreas tumor gyakoribb. Glucocerebrosidase.")
70
Gaucher-betegség II. Tipusai:
I. Tipus (=non-neuropathias): Askhenazi zsidók, késői gyerekkor vagy korai felnőttkor, élettartamot kissé csökkenti II. Tipus: Neurológiai problémák már csecsemőkorban III. Tipus: Svédország egyik régiójában gyakori Tünetek: Hepatomegalia, splenomegalia: -ott destrukciója a vvt, fvst, vérlemezkék →infekció, vérzés hajlam, anaemiaS Csontléziók (fájdalmas), osteoporosis, femur defomációja, nyirokcsomó duzzanat, sárgás-barnás árnyalata a bőrnek, sárga pigmentició a szemben Neurológiai: II. tipus: görcsök, mentális retardáció, hypertonia III. tipus: görcsök, dementia, szemozgató izmok bénulása
: Askhenazi zsidók, késői gyerekkor vagy. korai felnőttkor, élettartamot kissé csökkenti. II. Tipus: Neurológiai problémák már csecsemőkorban. III. Tipus: Svédország egyik régiójában gyakori. Tünetek: Hepatomegalia, splenomegalia: -ott destrukciója a vvt, fvst, vérlemezkék. →infekció, vérzés hajlam, anaemiaS. Csontléziók (fájdalmas), osteoporosis, femur defomációja, nyirokcsomó duzzanat, sárgás-barnás árnyalata a bőrnek, sárga pigmentició a szemben. Neurológiai: II. tipus: görcsök, mentális retardáció, hypertonia. III. tipus: görcsök, dementia, szemozgató izmok bénulása.")
71
Niemann-Pick-betegség I.
Condition involving the breakdown and use of fats and cholesterol in the body Harmful amounts of lipids accumulate in the spleen, liver, lungs, bone marrow, and brain Autosomal recessive pattern of inheritance (two copies of the gene must be present) Four variants: A, B, C1, and C2 Clinical feature include: severe liver disease, breathing difficulties, developmental delay, seizures, increased muscle tone, lack of coordination, problems feeding, and inability to move eyes vertically. No treatment
 Four variants: A, B, C1, and C2. Clinical feature include: severe liver disease, breathing difficulties, developmental delay, seizures, increased muscle tone, lack of coordination, problems feeding, and inability to move eyes vertically. No treatment.")
72
Variants Types A and B: mutated SMPD1 gene
SMPD gene carries instructions for cells to produce, sphingomyelinase, which processes lipids. Mutations lead to deficiency of sphingomyelinase and accumulations of cholesterol and lipids. Types C1 and C2: mutated NCP1 or NCP2 gene NCP1 gene produces a protein involved in the movement of cholesterol and lipids within a cell. May be a cholesterol pump, which is why its mutation leads to the buildup of lipids and cholesterol in the cell membrane. Plays a critical role in regulation of intracellular cholesterol trafficking NCP2 gene produces protein that binds and transports cholesterol (not fully understood).
.")
74
A génterápia diadala 1990. Az akkor 4 éves Ashanti DeSilva, aki génhiba miatt Súlyos Kombinált Immunhiányos Szindrómában (SCID) szenvedett. A hibátlan gént vírus segítségével juttatták a szervezetébe, és azóta tünetmentes. Az USA-ban jelenleg 200 génterápiás kezelést folytatnak klinikai kipróbálás céljából
 szenvedett. A hibátlan gént vírus segítségével juttatták a szervezetébe, és azóta tünetmentes. Az USA-ban jelenleg 200 génterápiás kezelést folytatnak klinikai kipróbálás céljából.")
75
és kudarca (egyben géndopping veszélye)
1998 September 17 Jesse Gelsinger. 18.-ként meghalt vírus allergia következtében, előtte 17-en meggyógyultak

Hasonló előadás












 MORE?” symposium Washington.>")







 n Áttekintés n Példák.>")

