Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
környezet Saját Non-self Veszélyes Patogén Immunrendszer Tolerancia Immunválasz The immunrendszer mint fekete doboz

2
Bacteriumok Virusok paraziták Vírus 3 óra Sokféleség Gyors fejlődés KÓROKOZÓK PATHOGENEK GENERACIÓS IDEJE RÖVID

3
Az emberek generációs ideje jóval hosszabb, fejlett immunrendszer kell 18 - 30 years

4
IMMUNODEFICIENCIÁK KOR ÉS EGÉSZSÉG FÜGGŐ IMMUNOSZUPPRESSZÍV GYÓGYSZEREK ÖRÖKLÖTT –Az immunrendszer génjeinek funkció vesztéses mutánsai –Fokozott érzékenység a fertőző betegségekkel szemben –Adott típusú patogénekkel szembeni érzékenység függően a gén defektustól –1950 óta ismerik, az antibiotikumok alkalmazása derített fényt a kórképek létezésére SZERZETT –Fertőző betegségek –AIDS –Más vírus fertőzések –Alultápláltság –Mesterséges immunszuppresszió Gyógyszerek Radioaktív besugárzás

5
ÖRÖKLÖTT IMMUNODEFICIENCIÁK A LEGTÖBB EGY ADOTT GÉN RECESSZÍV MUTÁCIÓJA –A domináns öröklött immundeficienciák eliminálódtak a populációból –Autoszomális gén defektus A betegség a homozigóta gyerekekben jelentkezik A heterozigóták hordozók –X-kromoszómához kapcsolt gének defektusai Egy gén defektusa a férfiakban betegséget okoz Egy gén defektusa a nőkben hordozóként jelentkezik Az IFNγ receptor mutációja a citokin kötés mellett az intracelluláris jelátvitel hiányát eredményezi - domináns Kontrollálatlan fertőzés a Mycobacterium ártalmatlan, vakcinációra használt törzsével szemben is (BCG oltás)
")
6
A recesszív és domináns IFN-γ receptor mutációk hatása monociták aktivációjára

7
Immunodeficiencia gének az X chromoszómán CGD: Krónikus Granulóma betegség WAS: Wiscott-Aldrich Syndrome SCID: Severe Combined Immunodeficiency XLA: X-linked Agammaglobulinemia XLP: X-linked Lymphoproliferative Disease XLHM: X-linked Hyper-IgM Syndrome

8
AZ ÖRÖKLÖTT IMMUNODEFICIENCIÁK TÍPUSAI B-SEJT DEFICIENCIA -Extracell. Bakt. fertőzés –B sejt fejlődés (XLA, IgA hiány) –B – T sejt együttműködés CD40 ligand, hiper IgM T SEJT DEFICIENCIA SCID, fertőzés opportunista patogénekkel –T sejt fejlődés IL-7/Jak3 RAG1/RAG2 mutációk –Tímusz epitél sejtek fejlődése DiGeorge szindróma –Purin lebontás zavara –DNS helyreállító enzim defektus –MHC II szintézis gátolt
 –B – T sejt együttműködés CD40 ligand, hiper IgM T SEJT DEFICIENCIA SCID, fertőzés opportunista patogénekkel –T sejt fejlődés IL-7/Jak3 RAG1/RAG2 mutációk –Tímusz epitél sejtek fejlődése DiGeorge szindróma –Purin lebontás zavara –DNS helyreállító enzim defektus –MHC II szintézis gátolt.")
9
KOMPLEMENT RENDSZER Extracelluláris bacteriális fertőzés Oldott és sejtfelszíni faktorok C3 C1 – C4 Komplement gátló faktorok FAGOCITA RENDSZER CD18 adhézió NADPH oxidáz Vezikuláris fúzió AZ ÖRÖKLÖTT IMMUNODEFICIENCIÁK TÍPUSAI

10
B-sejt eredetű Immunhiányos állapotok Kb 70%-a az összes ID-nak Későn manifesztálódik, 7-9 hónap Fokozott érzékenység: Tokos gennykeltő bakt. Streptococcus pneumoniae Haemophylus influenzae Enterovirusok, paraziták

11
Szérum Ig. szintek változása az egyedfejlődés során

12
Melyik sejttípus hiányzik? Extracelluláris baktériumok Haemophilus influentae Staphilococcus aureus Sterptococcus pyogenes

13
X-KROMOSZÓMÁHOZ KAPCSOLT AGAMMAGLOBULINEMIA XLA –Bruton agammaglobulinémia (1: 200 000) –Fokozott érzékenység a baktériumokkal és enterovírusokkal szemben antibiotikum –Gennykeltő baktériumok – Haemophilus influenzae, Streptococcus pyogenes, Staphylococcus aureus - folyamatos szövetkárosodás a bakteriális és makrofág eredetű enzimek miatt –bronchiectasis, krónikus tüdő betegség – havonta ismételt GG injekció vagy passzív ellenanyag terápia (egészséges egyedek plazmájából) –Kb 1000 szerumbol kevert (alacsony hőmérsékleten etanollal precipitalják, Edwin J Cohn, II. frakcio) –X-kromoszóma kapcsolt öröklésmenet –Fiú gyermek XY BETEG Fiú gyermek XY EGÉSZSÉGES –Mutáció a Bruton tirozin kináz (Btk) génben –A Btk B sejtekben, mielocitákban fejeződik ki –Elengedhetlen a B sejt aktivációhoz és fejlődéshez –NINCSENEK B SEJTEK A PERIFÉRIÁN – fejlődési blokk a pre-B szinten –Hordozó anya XX EGÉSZSÉGES nem random X inaktiváció B-sejtekben ELLENANYAG DEFICIENCIA AZ EXTRACELLULÁRIS BAKTÉRIUMOK NEM ELIMINÁLÓDNAK
 –X-kromoszóma kapcsolt öröklésmenet –Fiú gyermek XY BETEG Fiú gyermek XY EGÉSZSÉGES –Mutáció a Bruton tirozin kináz (Btk) génben –A Btk B sejtekben, mielocitákban fejeződik ki –Elengedhetlen a B sejt aktivációhoz és fejlődéshez –NINCSENEK B SEJTEK A PERIFÉRIÁN – fejlődési blokk a pre-B szinten –Hordozó anya XX EGÉSZSÉGES nem random X inaktiváció B-sejtekben ELLENANYAG DEFICIENCIA AZ EXTRACELLULÁRIS BAKTÉRIUMOK NEM ELIMINÁLÓDNAK.")
14
T-sejt eredetű immunhiány is okozhat csökkent ellenanyagtermelést. HIPER IgM SZINDRÓMA

15
–Defektus a CD40 ligand (CD40L) membrán citokin génben –X-kromoszómához kapcsolt, betegség férfiakban –Nincs specifikus ellenanyag válasz a T-dependens antigénekkel szemben Alacsony IgG, IgA, IgE –Nincs germinális centrum képződés –Súlyosabb minz az XLA---- MI AZ OKA??? –Nincs T sejt függő makrofág/DC/B sejt activáció a CD40 – CD40L kapcsolódás hiányában –Nincs gyulladás és leukocita mobilizáció –Nincs leukocitózis de tünete a neutropenia Gyulladás, hólyagok a szájban és a torokban GM-CSF injekció – monocita és granulocita pótlás –A gennykeltő baktériumokkal szemben fokozott érzékenység Antibiotikum Havi GG CSÖKKENT ELLENANYAG TERMELÉS A T SEJT SEGÍTSÉG KÁROSODÁSA MIATT

16
X-kapcsolt hiper IgM szindrómában szenvedő betegekben a Nyirokcsomókban nem alakulnak ki germinális centrumok

17
HYPER IgM Szindróma (Autoszómás) -Intrinsic B sejt hiba, az activation induced deaiminase (AID) hiánya okozza. Cytidine uridine reakciót katalizálja. -Az enzim szerepet játszik az ellenanyag affinitás érés és az izotípus váltás szabályozásában - nem jellemző az opportunista fertőzés megjelenése

18
SZELEKTÍV IgA DEFICIENCIA 1/800 Nincs általános fokozott érzékenység a fertőzésekkel szemben Krónikus tüdő betegség Allergiás reakciók (gasztrointesztínális rendszerben) Autoimmun folyamatok -Több mint 40%-a a betegeknek IgA-specifikus ellenanyagot termel – A vérkészítményekben lévő IgA súlyos allergiás reakcióhoz vezethet. - Egyes kórképek kapcsolatban vannak az MHC III régióval

19
T SEJT DEFICIENCIÁK

21
Candida albicans infection in children with SCID The Hart shadow is clearly visible In the absence of the thymus NormalSCID

22
Perzisztáló és ismétlődő fertőzések A patogének szélesebb típusaival szembeni érzékenység fokozódik Sem az ellenanyag, sem a celluláris immunválasz nem működik megfelelően A T SEJT FUNKCIÓK ZAVARAI A T sejtek az adaptív immunitás minden folyamatában részt vesznek SEVERE COMBINED IMMUNODEFICIENCY SCID SÚLYOS KOMBINÁLT IMMUNODEFICIENCIA Kezelés: Csontvelő átültetés, MHC-azonos testvér Gene therapy

23
X-SCID – Az interleukin receptorok közös γ-láncának hibája közös gamma lánc 55% totál, része az IL2,4,7,9, 15, 21 receptoroknak T- B+, NK-, a periférián a limfociták gyakorlatilag csak B sejtek Jak3 kináz mutációja IL-7 receptor jelátvitel SÚLYOS KOMBINÁLT IMMUNODEFICIENCIÁK A SCID fenotípust eltérő gén hibák okozhatják

24
Purin bázisok katabolizmusának hibája – autoszómás (T- B- NK+) –Adenosine deaminase (ADA) mutáció– –Purin nucleotide phosphorilase (PNP) purin metabolitok felhalmozódása Erősen toxikusak a fejlődő limfociták számára Autoszómás SCID A purin bázis lebomlás hibája ADA conc. A tímuszban kb 10-szeres

25
X-SCID – Az interleukin receptorok közös γ-láncának hibája közös gamma lánc 55% totál, része az IL2,4,7,9, 15, 21 receptoroknak T- B+, NK-, a periférián a limfociták gykorlatilag csak B sejtek Jak3 kináz mutációja IL-7 receptor jelátvitel RAG enzimek hibája –Omen szindróma T- B- SCID –Limfocita receptor génátrendeződés hiánya (gyorsan halálos) –Nincs T és B-sejt a periférián ha van szűk repertoire A purin bázis lebomlás hibája – autoszomális autoszómás (T- B- NK+) –Adenosine deaminase (ADA) mutáció – mentális retardáció –Purin nukleotid foszforiláz (PNP) Purin anyagcsere termékek halmozódása Erősen toxikus a fejlődő limfocitákra SÚLYOS KOMBINÁLT IMMUNODEFICIENCIÁK A SCID fenotípust eltérő gén hibák okozhatják
 –Nincs T és B-sejt a periférián ha van szűk repertoire A purin bázis lebomlás hibája – autoszomális autoszómás (T- B- NK+) –Adenosine deaminase (ADA) mutáció – mentális retardáció –Purin nukleotid foszforiláz (PNP) Purin anyagcsere termékek halmozódása Erősen toxikus a fejlődő limfocitákra SÚLYOS KOMBINÁLT IMMUNODEFICIENCIÁK A SCID fenotípust eltérő gén hibák okozhatják")
26
Kopasz limfocita szindróma BLS – MHC II szintézis gátolt –Nincs CD4+ T sejt válasz –CIITA ko-aktivátor, RFX promoter kötő fehérje vagy más transzkripciós hiba A DNS helyreállító enzimek hibája – autoszomális –DNS-függő protein kináz DiGeorge szindróma – a tímusz epitél sejtek fejlődésének hibája –T sejt fejlődés – pozitív szelekció károsodik –TAP transzporter hiba – a CD8+ T sejt válasz szelektív elvesztése – nem SCID fenotípus SÚLYOS KOMBINÁLT IMMUNODEFICIENCIÁK A SCID fenotípust eltérő gén hibák okozhatják

28
DiGeorge Syndrome Hypoparathyroidismus Thymus hypoplasia ami variábilis immundeficienciát okoz Egyéb jellegzetességek: Tipikus arc > 80% esetén a 22q11 kromoszóma deléciója Az érintett gén(ek) a T-box családba tartozó transzkripciós faktor Tbx1
 a T-box családba tartozó transzkripciós faktor Tbx1")
29
A B ésT sejt fejlődéshibái által okozott immundeficienciák

30
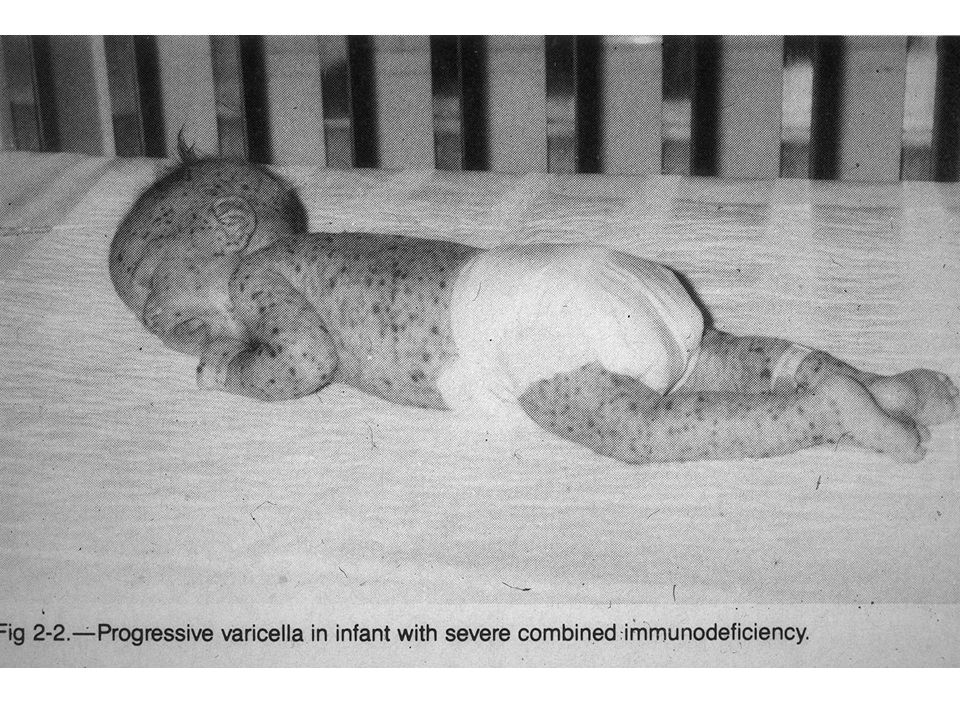
Wiskott-Aldrich szindroma WAS – X-kapcsolt A disease of defective reorganization of the actin cytoskeleton Tünetek: –Thrombocytopenia, kis méretű vérlemezkék, (csökkent termelés a csv-ben, fokozott elimináció a lépben) – Eczema –Alacsony IgM magas IgA, IgE szérum Ig szintek –Szénhidrát antigénekre specifikus IgM termelés csökkent mértékű (T sejtek szerepe?) –Súlyos varichella (bárányhimlő) fertőzés és herpes simplex (csökkent CD8+ T-sejt válasz) –Fertőzés tokos baktériumokkal, és opportunista fertőzések gyakoriak –B sejt lymphoma –Dinamikus aktin citoszkeleton átrendeződés, sejtpolarizáció defektív, T-B, T-Mfág CTL-Target interakció alkalmával Genetikai hiba –WAS protein (WASP) mutaciója fehérvérsejtekben és megakariocitákban funkciókiesést okoz Terápia Csontvelő átültetés
 – Eczema –Alacsony IgM magas IgA, IgE szérum Ig szintek –Szénhidrát antigénekre specifikus IgM termelés csökkent mértékű (T sejtek szerepe ) –Súlyos varichella (bárányhimlő) fertőzés és herpes simplex (csökkent CD8+ T-sejt válasz) –Fertőzés tokos baktériumokkal, és opportunista fertőzések gyakoriak –B sejt lymphoma –Dinamikus aktin citoszkeleton átrendeződés, sejtpolarizáció defektív, T-B, T-Mfág CTL-Target interakció alkalmával Genetikai hiba –WAS protein (WASP) mutaciója fehérvérsejtekben és megakariocitákban funkciókiesést okoz Terápia Csontvelő átültetés")
31
Wiskott-Aldrich szindroma WAS – X-kapcsolt Thrombocytopenia 40000 /μL + kisebb vérlemezkék Loss of microvilli on T cells in Wiskott–Aldrich syndrome. Scanning electron micrographs of normal lymphocytes (panel a) and lymphocytes from a patient with Wiskott– Aldrich syndrome (panel b). Note that the normal lymphocyte surface is covered with abundant microvilli, which are sparse or absent from the patient's lymphocytes. Photographs courtesy of Dianne Kenney.
 and lymphocytes from a patient with Wiskott– Aldrich syndrome (panel b). Note that the normal lymphocyte surface is covered with abundant microvilli, which are sparse or absent from the patient s lymphocytes. Photographs courtesy of Dianne Kenney..")
32
Wiskott-Aldrich syndrome WAS Hibás T/B Kommunikáció Fehér vérsejtekben, és megakariocitákban van jelen

33
Capping hiánya WASP negatív aktivált T sejtekben T sejtek WT egér T sejtek WASP-/- egér nyugvóanti CD3 kezelt

34
CD18 DEFICIENCIA/LEUKOCITA ADHÉZIÓ –A CR3, CR4 és LFA-1 közös β-alegysége –Gátolt fagocita migráció a vérből a fertőzés helyére –Az opszonizált baktériumok felvétele és lebontása gátolt –Perzisztáló fertőzések extracelluláris baktériumokkal Gennykeltő baktériumok A sebgyógyulás károsodása, súlyos íny gyulladás A FAGOCITA FUNKCIÓK KÁROSODÁAS FOKOZOTT ÉRZÉKENYSÉG A BAKTERIÁLIS FERTŐZÉSEKKEL SZEMBEN

35
Fontosabb adhéziós molekulák a leukocita interakciókban.

36
Omphalitis in LAD I patient CD18 DEFICIENCIA/LEUKOCITA ADHÉZIÓ HIÁNYA (LAD1) Rebuck bőrtesztí
 Rebuck bőrtesztí")
37
KRÓNIKUS GRANULÓMÁS BETEGSÉG - CGD A NADPH oxidáz mutációja – a 4 alegység bármelyike Gátolt NO és szuperoxid O2- gyök képződés az antibakteriális aktivitás gátolt, Aspergilus pneumonia Krónikus bakteriális fertőzések – granulóma képződés A glukóz-6-foszfát dehidrogenáz vagy mieloperoxidase gátolt működése kevéssé súlyos fenotípus A FAGOCITA FUNKCIÓK KÁROSODÁAS FOKOZOTT ÉRZÉKENYSÉG A BAKTERIÁLIS FERTŐZÉSEKKEL SZEMBEN CGD beteg Serratia Marcescens fertőzést követően granulómákkal

38
CHRONIC GRANULOMATOUS DISEASE – CGD NBT nitro-blue tetrazolium festés, neutr. granulociták Healthy CGD Hordozó Phox complex

39
C3 DEFICIENCIA VAGY GÁTOLT C3 AKTIVÁCIÓ –Érzékenység a gennykeltő baktériumokkal szemben –nem hatékony az opszonizáció C5- C9 deficiencia –Neisseria – NINCS komplement mediált lízis A KORAI C1-C4 KOMPONENSEK HIBÁJA Nem képződnek C3b és C4b fragmentumok Nem működik a CR1-mediált erythrocyta transzport és immunkomplex elimináció Immunkomplexek felhalmozódása a vérben, nyirokban, extracelluláris folyadékban lerakódás a szövetekben szövet károsodás makrofág aktiváció gyulladás OLDOTT ÉS MEMBRÁNHOZ KÖTÖTT KOMPLEMENT KOMPONENSEK HIBÁJÁBÓL ADÓDÓ IMMUNODEFICIENCIÁK

40
A KOMPLEMENTRENDSZER SZABÁLYOZÁSA

41
Hiányzó komplementfe hérje A hiány hatása C1, C2, C4 C3 Immunkomplex betegség (hasonlóan az SLE-hez), gennykeltő fertőzésekre való fogékonyság MAC, alternatív útvonal komponensek Fogékonyság Neisseria fertőzésre C1INHÖrökletes angioneurotikus ödéma (HANO) DAF (CD55), MIRL (CD59) Paroxysmalis nocturnalis haemoglobinuria (PNH) A komplementrendszer szabályozó molekuláinak és receptorainak deficienciái
, gennykeltő fertőzésekre való fogékonyság MAC, alternatív útvonal komponensek Fogékonyság Neisseria fertőzésre C1INHÖrökletes angioneurotikus ödéma (HANO) DAF (CD55), MIRL (CD59) Paroxysmalis nocturnalis haemoglobinuria (PNH) A komplementrendszer szabályozó molekuláinak és receptorainak deficienciái")
42
a PIG-A gén szerzett mutációja az őssejtekben, ennek következtében a hibás prekurzorokból származó sejtekben hiányoznak a membránban a GPI-horgonyzott fehérjék (klonális mutáció) ilyen GPI-horgonyzott fehérjék például a CD59 és a CD55 komplement szabályozó fehérjék ezek hiánya miatt a PNH betegek érintett sejtjei (vvs., thr., fvs.) hajlamosabbak a komplement- mediált lízisre anémia a vérsejtek lízise során felszabaduló hemoglobin bekerül a vizeletbe hemoglobinuria A károsodott leukocitákból szöveti faktor szabadul fel trombózisok Terápia: eculizumab (Soliris - C5 ellenes monoklonális antitest), csontvelőátültetés, szteroid PAROXYSMALIS NOCTURNALIS HEMOGLOBINURIA (PNH) PIG-A Phosphatidylinositol N-acetylglucosaminyl transferase Subunit A
 ilyen GPI-horgonyzott fehérjék például a CD59 és a CD55 komplement szabályozó fehérjék ezek hiánya miatt a PNH betegek érintett sejtjei (vvs., thr., fvs.) hajlamosabbak a komplement- mediált lízisre anémia a vérsejtek lízise során felszabaduló hemoglobin bekerül a vizeletbe hemoglobinuria A károsodott leukocitákból szöveti faktor szabadul fel trombózisok Terápia: eculizumab (Soliris - C5 ellenes monoklonális antitest), csontvelőátültetés, szteroid PAROXYSMALIS NOCTURNALIS HEMOGLOBINURIA (PNH) PIG-A Phosphatidylinositol N-acetylglucosaminyl transferase Subunit A")
43
PNH-S BETEGTŐL VETT VIZELETMINTÁK SZÍNÉNEK VÁLTOZÁSA A NAP SORÁN

44
HEREDITARY ANGIONEUROTIC EDEMA (HANE) (HEREDITARY C1INH DEFECT) Fő tünetek: bőr, belek, légutak duzzanata súlyos akut hasi fájdalom, hányás ödéma különféle helyeken (gégeduzzanat – fulladást okozhat) Kezelés: iv. C1INH, FFP, szteroid kallikrein inhibitor vagy bradikinin receptor antagonisták bradykinin és C2-kinin: megnövelik a posztkapilláris vénák permeabilitását ödema A plazmin jelenlétében a C1 folyamatosan hasítja a C2-őt és a C4-et Inhibition by C1INH in many steps

45
I faktor – nem kontrollált C3 C3b konverzió C3 depléció Properdin – a C3 lerakódás gátolt fokozott érzékenység Neisseria baktériummal szemben Decay Accelerating Factor DAF vagy a MIRL/CD59 MAC inhibitor – autoimmun jellegű állapot autológ erythrocyta lízis paroxysmalis nocturnalis hemoglobulinuria C1 inhibitor – a klasszikus út nem kontrollált aktivációja vazoaktív C2 folyadék felgyülemlés a szövetekben – epiglottális duzzanat, fulladásos halálhoz vezethez - Öröklött angioneurotikus ödema HANO KOMPLEMENT GÁTLÓ FAKTOROK HIBÁJA (SEJTFELSZÍNI)
")
46
C3 DEFICIENCIA VAGY GÁTOLT C3 AKTIVÁCIÓ –Érzékenység a gennykeltő baktériumokkal szemben –nem hatékony az opszonizáció C5-C9 deficiencia –Neisseria – NINCS komplement mediált lízis A KORAI C1-C4 KOMPONENSEK HIBÁJA –Nem képződnek C3b és C4b fragmentumok Nem működik a CR1-mediált erythrocyta transzport és immunkomplex elimináció –Immunkomplexek felhalmozódása a vérben, nyirokban, extracelluláris folyadékban lerakódás a szövetekben szövet károsodás makrofág aktiváció gyulladás KOMPLEMENT GÁTLÓ FAKTOROK HIBÁJA –I faktor – nem kontrollált C3 C3b konverzió C3 depléció –Properdin – a C3 lerakódás gátolt fokozott érzékenység Neisseria baktériummal szemben –Decay Accelerating Factor DAF vagy CD59 MAC inhibitor – autoimmun jellegű állapot autológ erythrocyta lízis paroxysmalis nocturnalis hemoglobulinuria –C1 inhibitor – a klasszikus út nem kontrollált aktivációja vazoaktív C2 folyadék felgyülemlés a szövetekben – epiglottális duzzanat, fulladásos halálhoz vezethez –Öröklött angioneurotikus ödema (HANO) A KOMPLEMENT KOMPONENSEK HIÁNYA NEM MEGFELELŐ AZ ELLENANYAG VÁLASZT EREDMÉNYEZ IMMUNKOMPLEXEK FELHALMOZÓDÁSA
 A KOMPLEMENT KOMPONENSEK HIÁNYA NEM MEGFELELŐ AZ ELLENANYAG VÁLASZT EREDMÉNYEZ IMMUNKOMPLEXEK FELHALMOZÓDÁSA")
Hasonló előadás
















 A szervezet sav-bázis egyensúlyának a felbomlása –”savasodás” A salakanyagok, nehézfémek,>")

 1/26 Energia és környezet NO x keletkezés és kibocsátás.>")

