Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
Cystás vesebetegségek diagnosztikája a genetika tükrében
Prof. Dr. Reusz György egyetemi tanár Semmelweis Egyetem I. sz. Gyermekklinika, Budapest

2
Cystás vesebetegségek diagnosztikája a genetika tükrében
Reusz György Tory Kálmán

3
Polycystás vesebetegségek
ARPKD ADPKD PKHD1 (génhordozás 1:65) 1:15.000 gyermekkorban CVE gyűjtőcsatorna… PKD1>PKD2 1: (5% sporadikus) A CVE 4. leggyakoribb oka CVE 50 (PKD1)-70 (PKD2) évesen gyűjtőcsatorna, distalis nephron…
 1: gyermekkorban CVE. gyűjtőcsatorna… PKD1>PKD2. 1: (5% sporadikus) A CVE 4. leggyakoribb oka. CVE 50 (PKD1)-70 (PKD2) évesen. gyűjtőcsatorna, distalis nephron…")
4
ARPKD A veseparenchyma helyét cysták foglalják el
ARPKD: Congenitalis májfibrosis A kóros fibrocystin protein (ld. később) a májat és a hasnyálmirígyet is érinti
 a májat és a hasnyálmirígyet is érinti.")
5
Nephronophthisis Tubulointerstitialis nephropathia
Cysták: a kéreg-velő határon, rendszerint kevés, nem igazi „cystás” vesebetegség! Klinikum Polyuria, polydypsia Anaemia (Normotensio) Veseelégtelenség 13 éves fiú, nephronophthisis 14 éves lány, nephronophthisis kontroll
 Veseelégtelenség. 13 éves fiú, nephronophthisis. 14 éves lány, nephronophthisis. kontroll.")
6
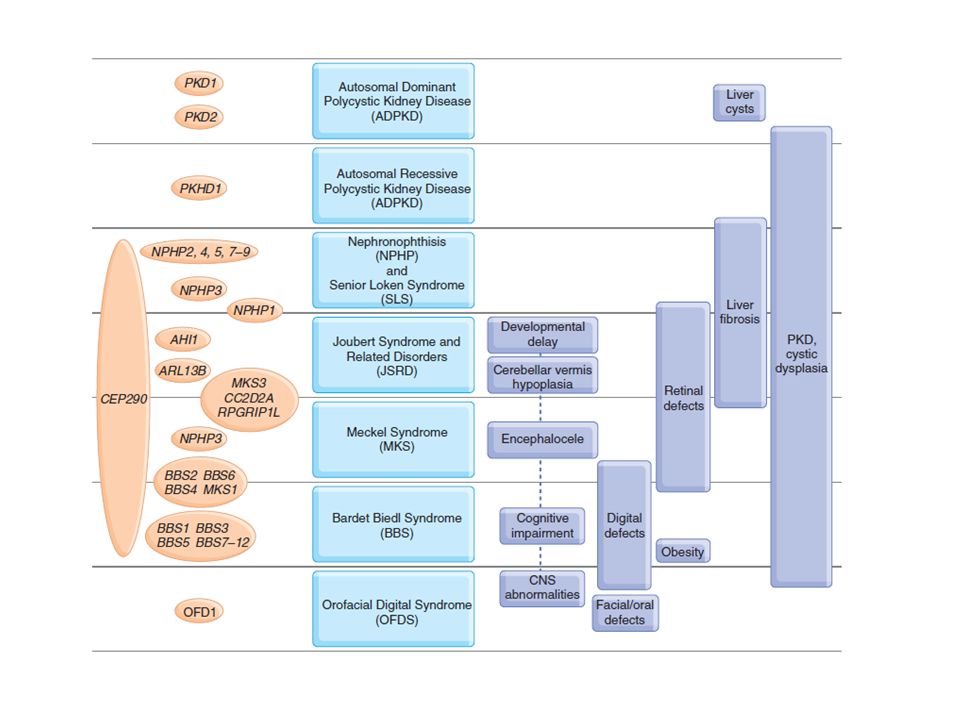
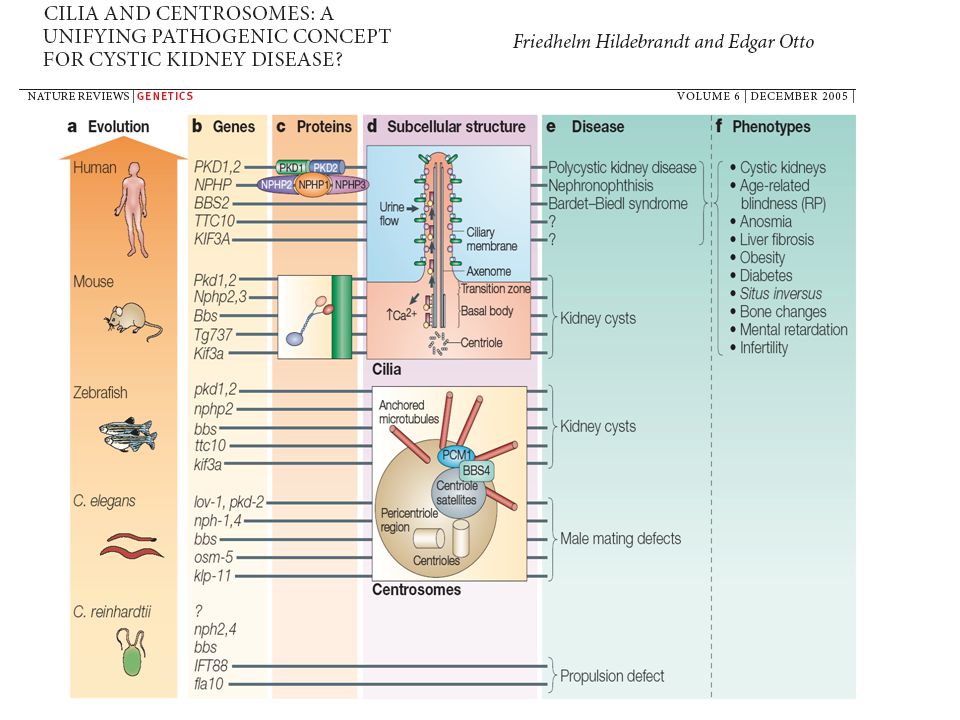
Örökletes cystás vesebetegségek
Közös pathomechanizmus?

7
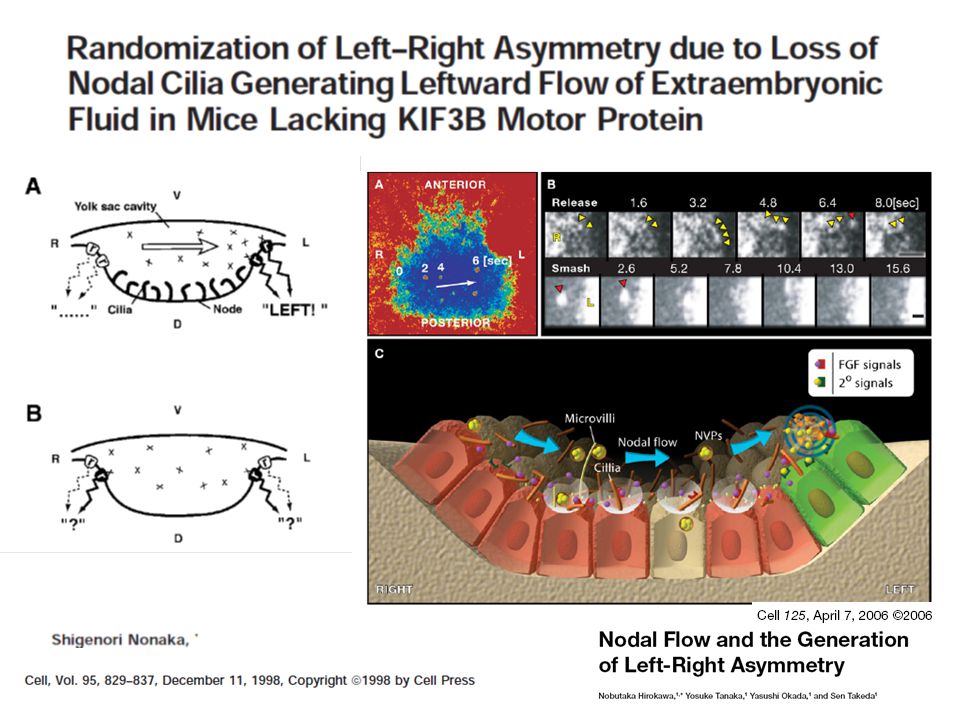
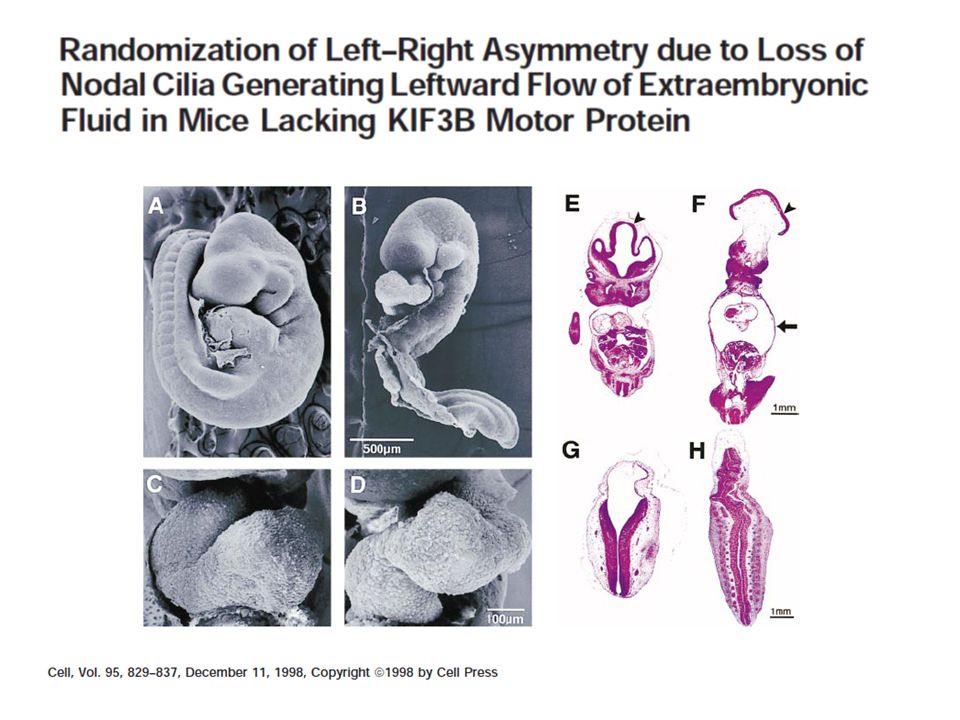
A fehérjék és a cilium történet
ADPKD A fehérjék és a cilium történet

8
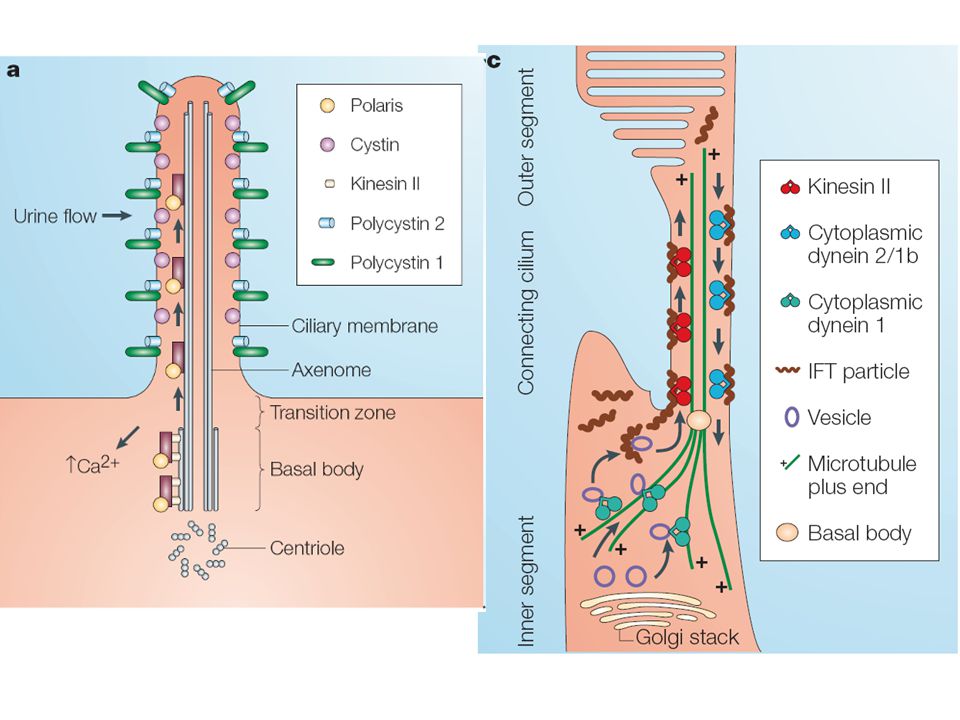
PKD1 polycystin1 PKD2 polycystin2 fibrocystin polyductin
Protein-protein kapcsolódás (decorin, biglycan, Toll receptor) Protein-cukor kapcs. (selectin) IgG- szerű (HGF receptor) Ligand kötődés, térbeli struktúra Ligand kötődés Acrosomális reakció során Ca beáramlást indukál Feszültség aktivált Ca/Na csatorna Tyrosin (SH 1-4) PKA-C target G protein akt. Coiled coil régió Tyrosin (SH 1-4) PKA-C target G protein akt. Coiled coil régió PKD1 polycystin1 PKD2 polycystin2 fibrocystin polyductin
 Protein-cukor. kapcs. (selectin) IgG- szerű. (HGF. receptor) Ligand kötődés, térbeli struktúra. Ligand kötődés. Acrosomális reakció során. Ca beáramlást indukál. Feszültség aktivált. Ca/Na csatorna. Tyrosin (SH 1-4) PKA-C target. G protein akt. Coiled coil régió. Tyrosin (SH 1-4) PKA-C target. G protein akt. Coiled coil régió. PKD1. polycystin1. PKD2. polycystin2. fibrocystin. polyductin.")
9
Polycystin-1 funkció: mechanoszenzor/kemoszenzor
1.5 milliárd éve konzervált funkciók Caenorhabditis elegans csillós érzékelő neuronok PKD1 és PKD2-t tartalmaznak acrosomalis reakció - Ca influx G protein jelátvitel szabályozása sejtproliferáció szabályozása/gátlása

10
Polycystin-2 50% homológia a polycystin-1-gyel
feszültség aktivált Ca csatorna COOH terminális régióval kapcsolódik a polycystin1-hez ER és sejtfelszíni lokalizáció

11
ARPKD 6p21.1-p12 régió 86 exon alternatív átírás/hasítás kb mRNS génproduktum: Polyductin/Fibrocystin sejtfelszíni receptor és/vagy szecernált protein, enzimaktivitással egér modell: egyes esetekben bal/jobb asszimetria hiánya

12
A mutációért felelős fehérjék csillókra lokalizálódnak
Annu. Rev. Physiol :83–113

13
PKD - a csillóbetegség? Tubulus sejteken 1-2 primér cilia
C. elegans polycystin-1 és -2 a csillós neuroszenzorra lokalizált egér mutánsokon PKD és bal/jobb asszimetria hiánya: polaris, cystin egyes ARPKD egérhomológokban is bal/jobb asszimetria hiánya

14
A csilló-koncepció kibontása: a nephronphthisis történet

15
Nephronophthisis Tubulointerstitialis nephropathia
Polyuria, polydypsia Anaemia (Normotensio) Veseelégtelenség További jellemzők Változó öröklésmenet, syndromatológia Joubert, Bardet Biedl Társuló anomáliák Máj Központi idegrendszer Retina Szaglás (Hallás) Situs inversus Kartagener syndroma
 Veseelégtelenség. További jellemzők. Változó öröklésmenet, syndromatológia. Joubert, Bardet Biedl. Társuló anomáliák. Máj. Központi idegrendszer. Retina. Szaglás. (Hallás) Situs inversus. Kartagener syndroma.")
16
Nephronophthisis Sokféleség … és ciliák

17
Cystic Diseases of the Kidney Molecular Biology and Genetics
Constantinos Deltas, PhD; Gregory Papagregoriou, MRes Arch Pathol Lab Med. 2010;134:569–582)
")
19
A csillók felépítése Annu. Rev. Physiol :83–113

20
egészséges mutáns

21
légzőrendszer primér csomó vese tubulus szaglóhám retina ívjárat

22
Egységesítő hypothesis
Az örökletes cystás betegségek oka: a ciliákat felépítő fehérjék, illetve az ahhoz kapcsolódó funkciók zavara Klinikai kép: függ attól, hogy az adott fehérje mely ciliákban fordul elő szöveti specificitás nephronophthisis átfedések situs inversus, Kartagener evolúciós példák

25
Ciliabetegség és vese: „iránytévesztés”
Egészséges PKD Sutter, JLCM 141:91, 2003 Annu. Rev. Physiol :83–113

26
A nephrocystinek - csillófehérjék
IFT-80 Les protéines codées par ces gènes sont les néphrocystines. On a entendu parler du rôle du cil primaire dans les maladies kystiques du rein. La plupart des protéines connues pour être mutées dans les maladies associés à la néphronophtise, comme la dystrophie thoracique, le syndrome de Joubert ou quelques protéines mutées dans la rétinopathie pigmentaire sont également localisées au cil primaire. Ceci peut expliquer pourquoi ces symptômes peuvent coexister chez un même patient. Nephrocystin 1,3,4,5,6, RPGR, RPGRIP1 Nephrocystin 1,6,8, meckelin

27
Extrarenális tünetek más gének episztatikus hatása miatt
NPHP1 homozigóta deléció 25% 75% + NPHP6, AHI1 heterozigóta mutáció Tory K et al. J Am Soc Nephrol 2007;18:

28
Ciliabetegség és retina: transzportzavar

30
Ciliabetegség és központi idegrendszer: „iránytévesztés”, migrációs zavar

31
Hypocampalis neuronok
Ciliabetegség és központi idegrendszer: „iránytévesztés”, migrációs zavar Hypocampalis neuronok sejttenyészetben Tubulus sejt Madeline A Lancaster: Current Opinion in Genetics & Development 2009, 19:220–229

33
Terápiás perspektívák

34
Intervenciós lehetőségek

35
Állatmodellek Humán Modell ARPKD ADPKD Pkd2WS25/- egér PCK patkány
cAMP cAMP

36
10 hetes PCK VP receptor génkiütött patkány
Hím Nőstény Wang. JASN, 2007

37
PCK patkányok VP antagonista (tolvaptan) kezelése
Kontroll Hím Nőstény Wang JASN 16:846, 2005

38
Metanephric organ culture
CFTR inhibítor terápia cAMP induced cystic Metanephric organ culture Forskolin induced MDCK cysts Control CFTR inh C172 Li: Kid Int 66:1926, 2004 Magenheimer JASN 17:3424, 2006

39
ADPKD és CYSTÁS FIBROSIS Enyhébb fenotípust eredményez
F508/ F508 O’Sullivan. AJKD 1998: 32: 976 Xu. J Nephrol 2006: 19: 529

40
ADPKD: Klinikai vizsgálatok
TEMPO (V2 RA, Otsuka) Octreotide (Bergamo, Mayo) Sirolimus (Cleveland, Zurich) Everolimus (Germany, Novartis) HALT-PKD (ACEI vs ACEI+ARB)
 Octreotide (Bergamo, Mayo) Sirolimus (Cleveland, Zurich) Everolimus (Germany, Novartis) HALT-PKD (ACEI vs ACEI+ARB)")
41
Összefoglalás A cystás vesebetegségek a csillókat alkotó fehérjék működésének zavara következtében alakulnak ki A struktúra komplexitása és szöveti/szervi eloszlása magyarázza a szerteágazó tüneteket A struktúra és funkció jobb megismerése megteremtheti a terápiás beavatkozás lehetőségét A hosszantartó terápia mellékhatásai ne haladják meg az alapbetegség ismert rizikóit

42
Nephronophthisis diagnosztika
Ismeretlen eredetű gyermekkori/fiatalkori veseelégtelenség („vesehypoplasia”) Jellegzetes tünetek: Tubulointerstitialis nephropathia Polyuria, polydypsia Anaemia (Normotensio) Veseelégtelenség Társuló szervi tünetek (máj, KIR, szem, situs inversus) Familiaritás genetikai diagnózis lehetősége az I. sz Gyermekklinikán
 Jellegzetes tünetek: Tubulointerstitialis nephropathia. Polyuria, polydypsia. Anaemia. (Normotensio) Veseelégtelenség. Társuló szervi tünetek (máj, KIR, szem, situs inversus) Familiaritás. genetikai diagnózis lehetősége az. I. sz Gyermekklinikán.")
44
A new spin on handed asymmetry Kyle J. Vogan and Clifford J. Tabin
NATURE|VOL 397 | 28 JANUARY 1999 |

45
Cystás vesebetegségek: Örökletes
Polycystás vesebetegség (AD, AR) Juvenilis nephronophthisis-medullaris cystás vese komplexum Multiplex fejlődési rendellenességgel társuló cysták Autosom domináns Sclerosis tuberosa von Hippel–Lindau betegség X kromoszómához kötött, domináns Orofaciodigitalis syndroma, I-es típus Autosom recesszív: Meckel syndroma, Jeune-féle mellkasi asphyxiás dystrophia, Zellweger cerebrohepatorenalis syndroma, Goldston syndroma, stb. (glomerulocystás vesebetegség az esetek zömében) Kromoszóma rendellenességek 21 trisomia, 13 trisomia, 18 trisomia
 Juvenilis nephronophthisis-medullaris cystás vese komplexum. Multiplex fejlődési rendellenességgel társuló cysták. Autosom domináns. Sclerosis tuberosa. von Hippel–Lindau betegség. X kromoszómához kötött, domináns. Orofaciodigitalis syndroma, I-es típus. Autosom recesszív: Meckel syndroma, Jeune-féle mellkasi asphyxiás dystrophia, Zellweger cerebrohepatorenalis syndroma, Goldston syndroma, stb. (glomerulocystás vesebetegség az esetek zömében) Kromoszóma rendellenességek. 21 trisomia, 13 trisomia, 18 trisomia.")
Hasonló előadás