Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
környezet Saját Non-self Veszélyes Patogén Immunrendszer Tolerancia Immunválasz The immunrendszer mint fekete doboz

2
Bacteriumok Virusok paraziták Vírus 3 óra Sokféleség Gyors fejlődés KÓROKOZÓK PATHOGENEK GENERACIÓS IDEJE RÖVID

3
Az emberek generációs ideje jóval hosszabb, fejlett immunrendszer kell 18 - 30 years

4
IMMUNODEFICIENCIÁK KOR ÉS EGÉSZSÉG FÜGGŐ IMMUNOSZUPPRESSZÍV GYÓGYSZEREK ÖRÖKLÖTT –Az immunrendszer génjeinek funkció vesztéses mutánsai –Fokozott érzékenység a fertőző betegségekkel szemben –Adott típusú patogénekkel szembeni érzékenység függően a gén defektustól –1950 óta ismerik, az antibiotikumok alkalmazása derített fényt a kórképek létezésére SZERZETT –Fertőző betegségek –AIDS –Más vírus fertőzések –Alultápláltság –Mesterséges immunszuppresszió Gyógyszerek Radioaktív besugárzás

5
ÖRÖKLÖTT IMMUNODEFICIENCIÁK A LEGTÖBB EGY ADOTT GÉN RECESSZÍV MUTÁCIÓJA –A domináns öröklött immundeficienciák eliminálódtak a populációból –Autoszomális gén defektus A betegség a homozigóta gyerekekben jelentkezik A heterozigóták hordozók –X-kromoszómához kapcsolt gének defektusai Egy gén defektusa a férfiakban betegséget okoz Egy gén defektusa a nőkben hordozóként jelentkezik Az IFNγ receptor mutációja a citokin kötés mellett az intracelluláris jelátvitel hiányát eredményezi - domináns Kontrollálatlan fertőzés a Mycobacterium ártalmatlan, vakcinációra használt törzsével szemben is (BCG oltás)
")
6
A recesszív és domináns IFN-γ receptor mutációk hatása monociták aktivációjára

7
Egyes génhibák hatására specifikus immundeficiencia alakulhat ki bizonyos típusú kórokozókkal szemben IL-12R-hiány

8
Immunodeficiencia gének az X chromoszómán CGD: Krónikus Granulóma betegség WAS: Wiscott-Aldrich Syndrome SCID: Severe Combined Immunodeficiency XLA: X-linked Agammaglobulinemia XLP: X-linked Lymphoproliferative Disease XLHM: X-linked Hyper-IgM Syndrome

9
AZ ÖRÖKLÖTT IMMUNODEFICIENCIÁK TÍPUSAI B-SEJT DEFICIENCIA -Extracell. Bakt. fertőzés –B sejt fejlődés (XLA, IgA hiány) –B – T sejt együttműködés CD40 ligand, hiper IgM T SEJT DEFICIENCIA SCID, fertőzés opportunista patogénekkel –T sejt fejlődés IL-7/Jak3 RAG1/RAG2 mutációk –Tímusz epitél sejtek fejlődése DiGeorge szindróma –Purin lebontás zavara –DNS helyreállító enzim defektus –MHC II szintézis gátolt
 –B – T sejt együttműködés CD40 ligand, hiper IgM T SEJT DEFICIENCIA SCID, fertőzés opportunista patogénekkel –T sejt fejlődés IL-7/Jak3 RAG1/RAG2 mutációk –Tímusz epitél sejtek fejlődése DiGeorge szindróma –Purin lebontás zavara –DNS helyreállító enzim defektus –MHC II szintézis gátolt.")
10
KOMPLEMENT RENDSZER Extracelluláris bacteriális fertőzés Oldott és sejtfelszíni faktorok C3 C1 – C4 Komplement gátló faktorok FAGOCITA RENDSZER CD18 adhézió NADPH oxidáz Vezikuláris fúzió AZ ÖRÖKLÖTT IMMUNODEFICIENCIÁK TÍPUSAI

11
B-sejt eredetű Immunhiányos állapotok Kb 70%-a az összes ID-nak Későn manifesztálódik, 7-9 hónap Fokozott érzékenység: Tokos gennykeltő bakt. Streptococcus pneumoniae Haemophylus influenzae Enterovirusok, paraziták

12
SERUM IG LEVELS AS THE DEVELOPMENT OF THE IMMUNE SYSTEM PROGRESSES

13
Melyik sejttípus hiányzik? Extracelluláris baktériumok Haemophilus influentae Staphilococcus aureus Sterptococcus pyogenes

14
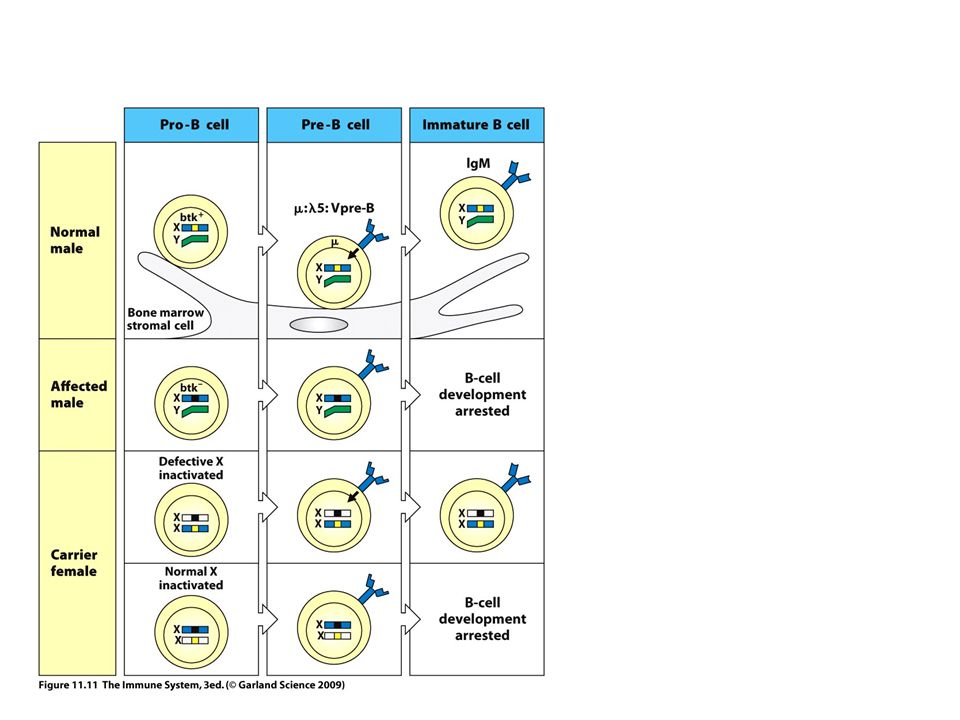
X-KROMOSZÓMÁHOZ KAPCSOLT AGAMMAGLOBULINEMIA XLA –Bruton agammaglobulinémia (1: 200 000) –Mutáció a Bruton tirozin kináz (Btk) génben –A Btk B sejtekben, mielocitákban fejeződik ki –Elengedhetlen a B sejt aktivációhoz és fejlődéshez –NINCSENEK B SEJTEK A PERIFÉRIÁN – fejlődési blokk a pre-B szinten –Hordozó anya XX EGÉSZSÉGES nem random X inaktiváció B-sejtekben –Fiú gyermek XY BETEG Fiú gyermek XY EGÉSZSÉGES –Fokozott érzékenység a baktériumokkal és enterovírusokkal szemben antibiotikum –Gennykeltő baktériumok – folyamatos szövetkárosodás a bakteriális és makrofág eredetű enzimek miatt –bronchiectasis, krónikus tüdő betegség – havonta ismételt GG injekció vagy passzív ellenanyag terápia (egészsége egyedek plazmájából) ELLENANYAG DEFICIENCIA AZ EXTRACELLULÁRIS BAKTÉRIUMOK NEM ELIMINÁLÓDNAK
 –Mutáció a Bruton tirozin kináz (Btk) génben –A Btk B sejtekben, mielocitákban fejeződik ki –Elengedhetlen a B sejt aktivációhoz és fejlődéshez –NINCSENEK B SEJTEK A PERIFÉRIÁN – fejlődési blokk a pre-B szinten –Hordozó anya XX EGÉSZSÉGES nem random X inaktiváció B-sejtekben –Fiú gyermek XY BETEG Fiú gyermek XY EGÉSZSÉGES –Fokozott érzékenység a baktériumokkal és enterovírusokkal szemben antibiotikum –Gennykeltő baktériumok – folyamatos szövetkárosodás a bakteriális és makrofág eredetű enzimek miatt –bronchiectasis, krónikus tüdő betegség – havonta ismételt GG injekció vagy passzív ellenanyag terápia (egészsége egyedek plazmájából) ELLENANYAG DEFICIENCIA AZ EXTRACELLULÁRIS BAKTÉRIUMOK NEM ELIMINÁLÓDNAK")
16
T-sejt eredetű immunhiány is okozhat csökkent ellenanyagtermelést. HIPER IgM SZINDRÓMA

17
–Defektus a DC40 ligand (CD40L) membrán citokin génben –X-kromoszómához kapcsolt, betegség férfiakban –Nincs specifikus ellenanyag válasz a T-dependens antigénekkel szemben Alacsony IgG, IgA, IgE –Nincs germinális centrum képződés –Nincs T sejt függő makrofág/DC/B sejt activáció a CD40 – CD40L kapcsolódás hiányában –Nincs gyulladás és leukocita mobilizáció –Nincs leukocitózis de tünete a neutropenia Gyulladás, hólyagok a szájban és a torokban GM-CSF injekció – monocita és granulocita pótlás –A gennykeltő baktériumokkal szemben fokozott érzékenység Antibiotikum Havi GG CSÖKKENT ELLENANYAG TERMELÉS A T SEJT SEGÍTSÉG KÁROSODÁSA MIATT
 membrán citokin génben –X-kromoszómához kapcsolt, betegség férfiakban –Nincs specifikus ellenanyag válasz a T-dependens antigénekkel szemben Alacsony IgG, IgA, IgE –Nincs germinális centrum képződés –Nincs T sejt függő makrofág/DC/B sejt activáció a CD40 – CD40L kapcsolódás hiányában –Nincs gyulladás és leukocita mobilizáció –Nincs leukocitózis de tünete a neutropenia Gyulladás, hólyagok a szájban és a torokban GM-CSF injekció – monocita és granulocita pótlás –A gennykeltő baktériumokkal szemben fokozott érzékenység Antibiotikum Havi GG CSÖKKENT ELLENANYAG TERMELÉS A T SEJT SEGÍTSÉG KÁROSODÁSA MIATT")
18
X-kapcsolt hiper IgM szindrómában szenvedő betegekben a Nyirokcsomókban nem alakulnak ki germinális centrumok

19
HYPER IgM Szindróma (Autoszómás) -Intrinsic B sejt hiba, az activation induced deaiminase (AID) hiánya okozza. Cytidine uridine reakciót katalizálja. -Az enzim szerepet játszik az affinitás érés és az izotípus váltás szabályozásában - nem jellemző az opportunista fertőzés megjelenése

20
Szelektív IgA deficiencia Gyakoriság: 1:160-1:800 (Európában, Ázsiában ritkább) -Tünetmentes is lehet de -Gyakori vagy krónikus légúti fertőzés (tüdő is ) előfordul, -Allergiás tünetek -Girardia fertőzés gyakoribb, kronikus hasmenés -Autoimmun betegség, autoimmun citopénia - A betegek kb. 40%-a termel anti-IgA antitestet amennyiben IgA tartalmu vérkészítményt kaptak.

21
CVID, Common variable Immunodeficiency Gyakoriság: 1:25000 Tünetek: Alacsony IgG szint alacsony IgAés/vagy IgM Gyakori fertőzések légúti, fül, orr Granulómás fertőzések a tüdőben és a bélrendszerben, hasmenés Gyenge immunválasz vakcinációra Csak 10 % -körül van család-története a betegségnek 10%-körül ismert a génhiba Autoimmun citopénia Hematológiai eredetű daganatos betegségek, limfóma IVIG terápia Számos esetben megtalálják a hibás gént Inducible costimulator (ICOS) Transmembrane activator and calcium modulator and cyclophilin ligand (TACI) BAFF- Receptor
 Transmembrane activator and calcium modulator and cyclophilin ligand (TACI) BAFF- Receptor")
22
T SEJT DEFICIENCIÁK

24
Perzisztáló és ismétlődő fertőzések A patogének szélesebb típusaival szembeni érzékenység fokozódik Sem az ellenanyag, sem a celluláris immunválasz nem működik megfelelően A T SEJT FUNKCIÓK ZAVARAI A T sejtek az adaptív immunitás minden folyamatában részt vesznek SEVERE COMBINED IMMUNODEFICIENCY SCID SÚLYOS KOMBINÁLT IMMUNODEFICIENCIA Kezelés: Csontvelő átültetés, MHC-azonos testvér Gene therapy

25
Néhány súlyos kombinált immundeficienciát okozó mutáció

26
X-SCID – Az interleukin receptorok közös γ-láncának hibája közös gamma lánc 55% totál, része az IL2,4,7,9, 15, 21 receptoroknak T- B+, NK-, a periférián a limfociták gykorlatilag csak B sejtek Jak3 kináz mutációja IL-7 receptor jelátvitel RAG enzimek hibája –Omen szindróma T- B- SCID –Limfocita receptor génátrendeződés hiánya (gyorsan halálos) –Nincs T és B-sejt a periférián ha van szűk repertoire A purin bázis lebomlás hibája – autoszomális autoszómás (T- B- NK+) –Adenosine deaminase (ADA) mutáció – mentális retardáció –Purin nukleotid foszforiláz (PNP) Purin anyagcsere termékek halmozódása Erősen toxikus a fejlődő limfocitákra Kopasz limfocita szindróma BLS – MHC II szintézis gátolt –Nincs CD4+ T sejt válasz –CIITA ko-aktivátor, RFX promoter kötő fehérje vagy más transzkripciós hiba A DNS helyreállító enzimek hibája – autoszomális –DNS-függő protein kináz (DNA-PK) DiGeorge szindróma – a tímusz epitél sejtek fejlődésének hibája –T sejt fejlődés – pozitív szelekció károsodik SÚLYOS KOMBINÁLT IMMUNODEFICIENCIÁK A SCID fenotípust eltérő gén hibák okozhatják
 –Nincs T és B-sejt a periférián ha van szűk repertoire A purin bázis lebomlás hibája – autoszomális autoszómás (T- B- NK+) –Adenosine deaminase (ADA) mutáció – mentális retardáció –Purin nukleotid foszforiláz (PNP) Purin anyagcsere termékek halmozódása Erősen toxikus a fejlődő limfocitákra Kopasz limfocita szindróma BLS – MHC II szintézis gátolt –Nincs CD4+ T sejt válasz –CIITA ko-aktivátor, RFX promoter kötő fehérje vagy más transzkripciós hiba A DNS helyreállító enzimek hibája – autoszomális –DNS-függő protein kináz (DNA-PK) DiGeorge szindróma – a tímusz epitél sejtek fejlődésének hibája –T sejt fejlődés – pozitív szelekció károsodik SÚLYOS KOMBINÁLT IMMUNODEFICIENCIÁK A SCID fenotípust eltérő gén hibák okozhatják")
27
Purin bázisok katabolizmusának hibája – autoszómás (T- B- NK+) –Adenosine deaminase (ADA) mutáció– –Purin nucleotide phosphorilase (PNP) purin metabolitok felhalmozódása Erősen toxikusak a fejlődő limfociták számára Autoszómás SCID ADA conc. A tímuszban kb 10-szeres

28
Diagnózis? Leggyakrabban a súlyosan beteg SCID beteg kerül a klinikára „rendbe kell hozni” csontvelő átültetés előtt Lehet-e a betegség kialakulása előtt azonosítani a SCID-eket??? SCID Newborn Screening Campaign: On January 21, 2010, the Advisory Committee on Heritable Disorders in Newborns and Children voted unanimously to add screening for Severe Combined Immune Deficiency or SCID – commonly known as bubble boy disease – to the core panel for universal screening of all newborns in the United States. On May 21, 2010 Kathleen Sebelius, Secretary of Health and Services announced the addition of Severe Combined Immunodeficiency (SCID) to the core panel of 29 genetic disorders as part of her recommendation to adopt the national Recommended Uniform Screening Panel. SCID is the first nominated condition to be added to the core panel of disorders. States and US Territories screening all newborns for SCID are: WI, MA, NY, CA, CT, MI, CO, MS, DE, FL, TX, MN, IA, PA, UT and OH. T-cell receptor excision circle assay has revolutionized early identification of infants with SCID or severe T-cell lymphopenia. TREC fragments are identified by QPCR
 to the core panel of 29 genetic disorders as part of her recommendation to adopt the national Recommended Uniform Screening Panel. SCID is the first nominated condition to be added to the core panel of disorders. States and US Territories screening all newborns for SCID are: WI, MA, NY, CA, CT, MI, CO, MS, DE, FL, TX, MN, IA, PA, UT and OH. T-cell receptor excision circle assay has revolutionized early identification of infants with SCID or severe T-cell lymphopenia. TREC fragments are identified by QPCR.")
29
Loop of intervening DNA is excised V1 DJ V2 V3 V4 V8 V7 V6 V5 V9 A rekombináció alkalmával T-cell receptor excision circle képződik A TREC QPCR-rel detektálható Korai diagnózis Gyakorlatilag nincs fals pozitív

31
DiGeorge Syndrome Hypoparathyroidismus Thymus hypoplasia ami variábilis immundeficienciát okoz Egyéb jellegzetességek: Tipikus arc > 80% esetén a 22q11 kromoszóma deléciója Az érintett gén(ek) a T-box családba tartozó transzkripciós faktor Tbx1
 a T-box családba tartozó transzkripciós faktor Tbx1")
33
Wiskott-Aldrich szindroma WAS – X-kapcsolt A disease of defective reorganization of the actin cytoskeleton Tünetek: –Thrombocytopenia, kis méretű vérlemezkék, (csökkent termelés a csv-ben, fokozott elimináció a lépben) – Eczema –Alacsony IgM magas IgA, IgE szérum Ig szintek –Szénhidrát antigénekre specifikus IgM termelés csökkent mértékű (T sejtek szerepe?) –Súlyos varichella (bárányhimlő) fertőzés és herpes simplex (csökkent CD8+ T-sejt válasz) –Fertőzés tokos baktériumokkal, és opportunista fertőzések gyakoriak –B sejt lymphoma –Dinamikus aktin citoszkeleton átrendeződés, sejtpolarizáció defektív, T-B, T-Mfág CTL-Target interakció alkalmával Genetikai hiba –WAS protein (WASP) mutaciója fehérvérsejtekben és megakariocitákban funkciókiesést okoz Terápia Csontvelő átültetés
 – Eczema –Alacsony IgM magas IgA, IgE szérum Ig szintek –Szénhidrát antigénekre specifikus IgM termelés csökkent mértékű (T sejtek szerepe ) –Súlyos varichella (bárányhimlő) fertőzés és herpes simplex (csökkent CD8+ T-sejt válasz) –Fertőzés tokos baktériumokkal, és opportunista fertőzések gyakoriak –B sejt lymphoma –Dinamikus aktin citoszkeleton átrendeződés, sejtpolarizáció defektív, T-B, T-Mfág CTL-Target interakció alkalmával Genetikai hiba –WAS protein (WASP) mutaciója fehérvérsejtekben és megakariocitákban funkciókiesést okoz Terápia Csontvelő átültetés")
34
Wiskott-Aldrich syndrome WAS Hibás T/B Kommunikáció Fehér vérsejtekben, és megakariocitákban van jelen

35
Capping hiánya WASP negatív aktivált T sejtekben T sejtek WT egér T sejtek WASP-/- egér nyugvóanti CD3 kezelt

36
CD18 DEFICIENCIA/LEUKOCITA ADHÉZIÓ –A CR3, CR4 és LFA-1 közös β-alegysége –Gátolt fagocita migráció a vérből a fertőzés helyére –Az opszonizált baktériumok felvétele és lebontása gátolt –Perzisztáló fertőzések extracelluláris baktériumokkal Gennykeltő baktériumok A sebgyógyulás károsodása, súlyos íny gyulladás A FAGOCITA FUNKCIÓK KÁROSODÁAS FOKOZOTT ÉRZÉKENYSÉG A BAKTERIÁLIS FERTŐZÉSEKKEL SZEMBEN

37
Omphalitis in LAD I patient CD18 DEFICIENCIA/LEUKOCITA ADHÉZIÓ HIÁNYA (LAD1)
")
38
KRÓNIKUS GRANULÓMÁS BETEGSÉG - CGD A NADPH oxidáz mutációja – a 4 alegység bármelyike Gátolt NO és szuperoxid O2- gyök képződés az antibakteriális aktivitás gátolt, Aspergilus pneumonia Krónikus bakteriális fertőzések – granulóma képződés A glukóz-6-foszfát dehidrogenáz vagy mieloperoxidase gátolt működése kevéssé súlyos fenotípus A FAGOCITA FUNKCIÓK KÁROSODÁAS FOKOZOTT ÉRZÉKENYSÉG A BAKTERIÁLIS FERTŐZÉSEKKEL SZEMBEN CGD beteg Serratia Marcescens fertőzést követően granulómákkal

39
CHRONIC GRANULOMATOUS DISEASE – CGD NBT festés, neutr. granulociták Healthy CGD Hordozó Phox complex

40
CHÉDIAK-HIGASHI SYNDROME Abnormal giant granules in a variety of cells leading to: -hypopigmentation/partial albinism hair and eyes -severe immunodeficiency - NK cell defect Defect in vesicle fusion mechanism phagocytosed material is not delivered to lysosomes Persistent and recurrent bacterial infections Patients with CHS exhibit hypopigmentation of the skin, eyes, and hair; prolonged bleeding times; large bruises; recurrent infections; abnormal natural killer cell function; Infections most commonly involve the skin, the lungs, and the respiratory tract and are usually due to Staphylococcus aureus, Streptococcus pyogenes, and Pneumococcus species. Defective melanization of melanosomes occurs in oculocutaneous albinism In melanocytes, autophagocytosis of melanosomes occurs. Defective gene: CHS1 located on 1q42-43 DEFECTS IN PHAGOCYTE FUNCTION ENHANCED SUSCEPTIBILITY TO BACTERIAL INFECTIONS

41
A fagocita sejtek defektusai perzisztens bakteriális fertőzéseket okoznak

42
C3 DEFICIENCIA VAGY GÁTOLT C3 AKTIVÁCIÓ –Érzékenység a gennykeltő baktériumokkal szemben –nem hatékony az opszonizáció C5-C9 deficiencia –Neisseria – NINCS komplement mediált lízis A KORAI C1-C4 KOMPONENSEK HIBÁJA –Nem képződnek C3b és C4b fragmentumok Nem működik a CR1-mediált erythrocyta transzport és immunkomplex elimináció –Immunkomplexek felhalmozódása a vérben, nyirokban, extracelluláris folyadékban lerakódás a szövetekben szövet károsodás makrofág aktiváció gyulladás KOMPLEMENT GÁTLÓ FAKTOROK HIBÁJA –I faktor – nem kontrollált C3 C3b konverzió C3 depléció –Properdin – a C3 lerakódás gátolt fokozott érzékenység Neisseria baktériummal szemben –Decay Accelerating Factor DAF vagy CD59 MAC inhibitor – autoimmun jellegű állapot autológ erythrocyta lízis paroxysmalis nocturnalis hemoglobulinuria –C1 inhibitor – a klasszikus út nem kontrollált aktivációja vazoaktív C2 folyadék felgyülemlés a szövetekben – epiglottális duzzanat, fulladásos halálhoz vezethez –Öröklött angioneurotikus ödema (HANO) A KOMPLEMENT KOMPONENSEK HIÁNYA NEM MEGFELELŐ AZ ELLENANYAG VÁLASZT EREDMÉNYEZ IMMUNKOMPLEXEK FELHALMOZÓDÁSA
 A KOMPLEMENT KOMPONENSEK HIÁNYA NEM MEGFELELŐ AZ ELLENANYAG VÁLASZT EREDMÉNYEZ IMMUNKOMPLEXEK FELHALMOZÓDÁSA")
43
A KORAI C1-C4 KOMPONENSEK HIBÁJA Nem képződnek C3b és C4b fragmentumok Nem működik a CR1-mediált erythrocyta transzport és immunkomplex elimináció Immunkomplexek felhalmozódása a vérben, nyirokban, extracelluláris folyadékban lerakódás a szövetekben szövet károsodás makrofág aktiváció gyulladás OLDOTT ÉS MEMBRÁNHOZ KÖTÖTT KOMPLEMENT KOMPONENSEK HIBÁJÁBÓL ADÓDÓ IMMUNODEFICIENCIÁK

44
I faktor – nem kontrollált C3 C3b konverzió C3 depléció Properdin – a C3 lerakódás gátolt fokozott érzékenység Neisseria baktériummal szemben Decay Accelerating Factor DAF vagy a MIRL/CD59 MAC inhibitor – autoimmun jellegű állapot autológ erythrocyta lízis paroxysmalis nocturnalis hemoglobulinuria C1 inhibitor – a klasszikus út nem kontrollált aktivációja vazoaktív C2 folyadék felgyülemlés a szövetekben – epiglottális duzzanat, fulladásos halálhoz vezethez - Öröklött angioneurotikus ödema HANO KOMPLEMENT GÁTLÓ FAKTOROK HIBÁJA (SEJTFELSZÍNI)
")
45
X-linked Lymphoproliferative disease XLP

46
Symptoms of XLP 1.Fulminant Mononukleózis (57 %) Masszív limfocita proliferáció és citokin termelés (IFN-γ, TNF, IL-1, IL-2) Szöveti károsodás, több szerv együttes funkció kiesése (máj, csontvelő, tímusz) (Timocita depléció, timusz epitélium pusztulása) Vasculitis, erythrocyte phagocytosis í8aktivált makrofágok által) Aplastic anemia, thrombocytopenia Cytokin-indukált Fas-expresszió májsekteken Szeptikus sokk szerű tünetek: D examethasone and etoposide, vagy anti-CD20 antitest 2. B-sejt lymphoma (20 %) EBV jelenlétében és hiányában is 3. Hypo- vagy agammaglobulinémia (30 %) EBV jelenlétében és hiányában is 1975. David Purtilo
 EBV jelenlétében és hiányában is 3. Hypo- vagy agammaglobulinémia (30 %) EBV jelenlétében és hiányában is David Purtilo.")
47
XLP: A SAP/SH2D1A hiány Ma et al Ann Rev. Immunol 2007

Hasonló előadás

















>")
>")

 kialakulása Genetikai, Sejt- és Immunbiológiai Intézet Falus András.>")

