Előadást letölteni
1
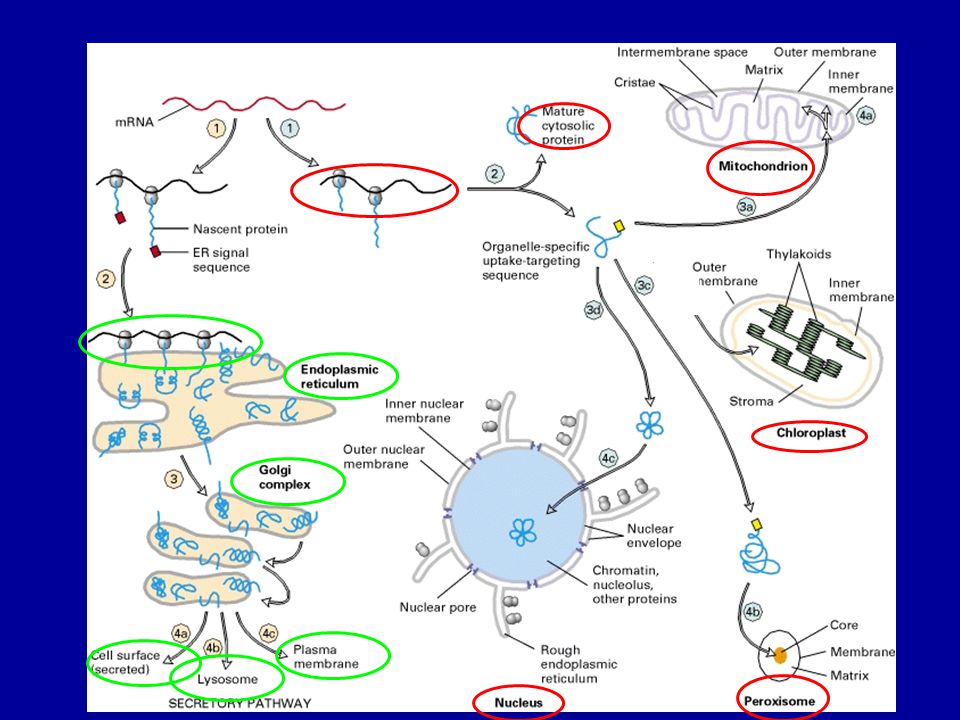
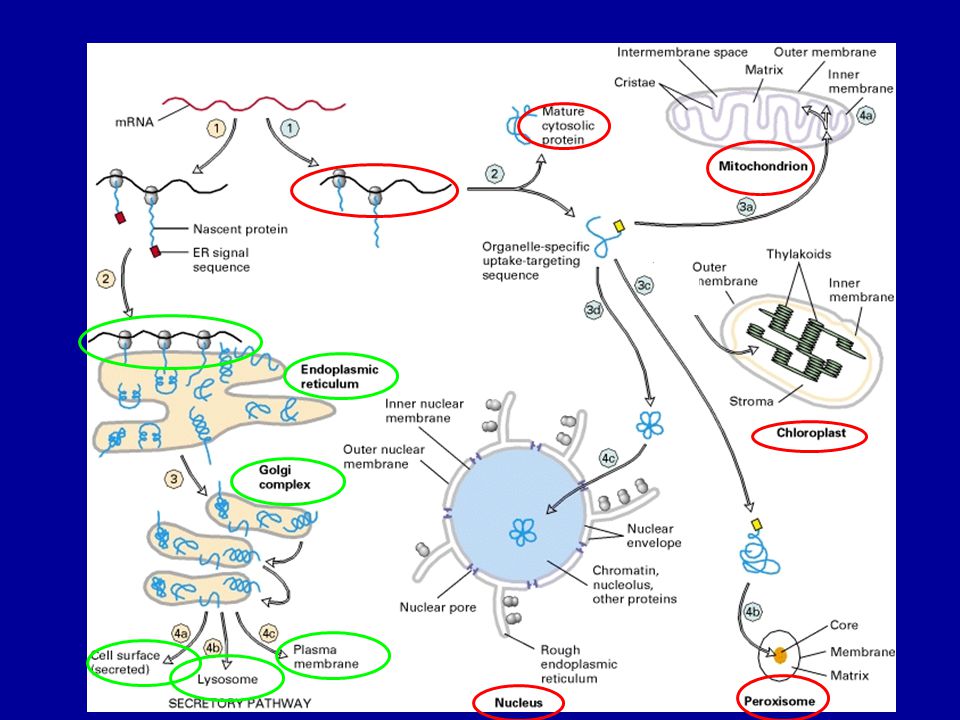
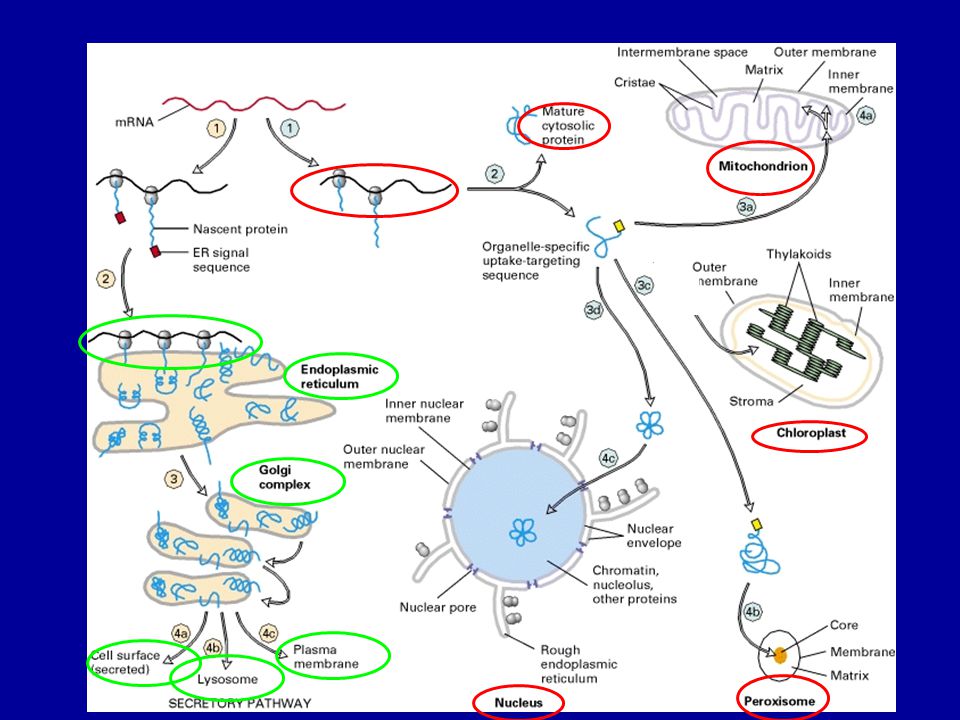
A sejten belüli fehérje
transzport mechanizmusok

3
Mi történik a fehérjékkel az ER-ben?
Kotranszlációs transzmembrán transzport Transzmembrán fehérjék végleges orientációja Proteolízis (szignál peptidáz) N-glikoziláció és módosítás (szintézis közben) Hajtogatás (PDI –protein diszulfid izomeráz, calnexin, calretikulin, BiP) Multimer protein összeszerelése Rosszul hajtogatott fehérjék citoszolba történő transzportja Cél: funkcióképes térszerkezet (harmadlagos, negyedleges) kialakítása = Minőség ellenőrzés
 N-glikoziláció és módosítás (szintézis közben) Hajtogatás (PDI –protein diszulfid izomeráz, calnexin, calretikulin, BiP) Multimer protein összeszerelése. Rosszul hajtogatott fehérjék citoszolba történő transzportja. Cél: funkcióképes térszerkezet. (harmadlagos, negyedleges) kialakítása = Minőség ellenőrzés.")
4
ER lumen fő jellemzői: oxidatív magas Ca2+

5
Minőség ellenőrzés az ER-ben

6
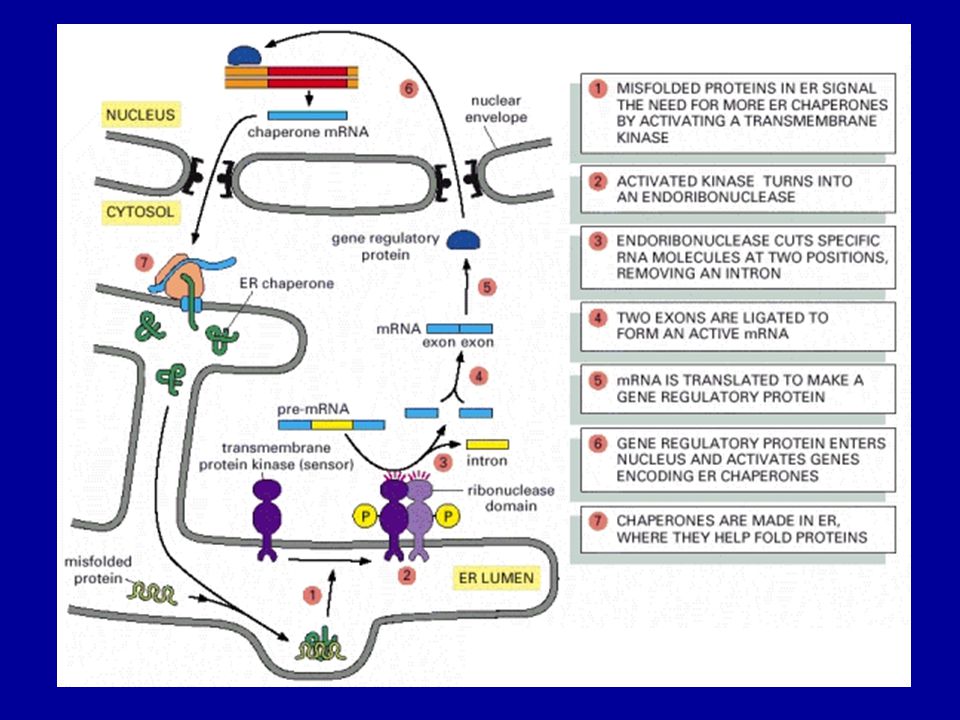
he ER stress response pathway (7)
he ER stress response pathway (7). Accumulation of unfolded proteins in the ER activates four distinct cellular responses. (A) Transcriptional induction of ER chaperones increases protein folding activity and prevents protein aggregation. (B) Translational attenuation reduces the load of new protein synthesis and prevents further accumulation of unfolded proteins. (C) The ER-associated degradation (ERAD) pathway eliminates misfolded proteins by the ubiquitin-proteasome system. (D) When functions of the ER are severely impaired, apoptosis is induced to destroy the cell.
. Accumulation of unfolded proteins in the ER activates four distinct cellular responses. (A) Transcriptional induction of ER chaperones increases protein folding activity and prevents protein aggregation. (B) Translational attenuation reduces the load of new protein synthesis and prevents further accumulation of unfolded proteins. (C) The ER-associated degradation (ERAD) pathway eliminates misfolded proteins by the ubiquitin-proteasome system. (D) When functions of the ER are severely impaired, apoptosis is induced to destroy the cell.")
7
Protein degradáció a proteaszómában
poli

8
Unfolded protein response (UPR)
Csaperon expresszió A rosszul hajtogatott fehérjék indukálják a hajtogatást segítő csaperonok szintézisét.

10
Endoplazmás retikulum tárolási
betegségek (ERSD)
")
11
A fehérje hiánya okozza a betegséget
– funkció vesztés (loss of function) A felhalmozódó hibás fehérje okozza a betegséget - toxikus funkció nyerés (gain of toxic function)
 A felhalmozódó hibás fehérje okozza a betegséget. - toxikus funkció nyerés (gain of. toxic function)")
12
Endoplazmás retikulum tárolási betegségek
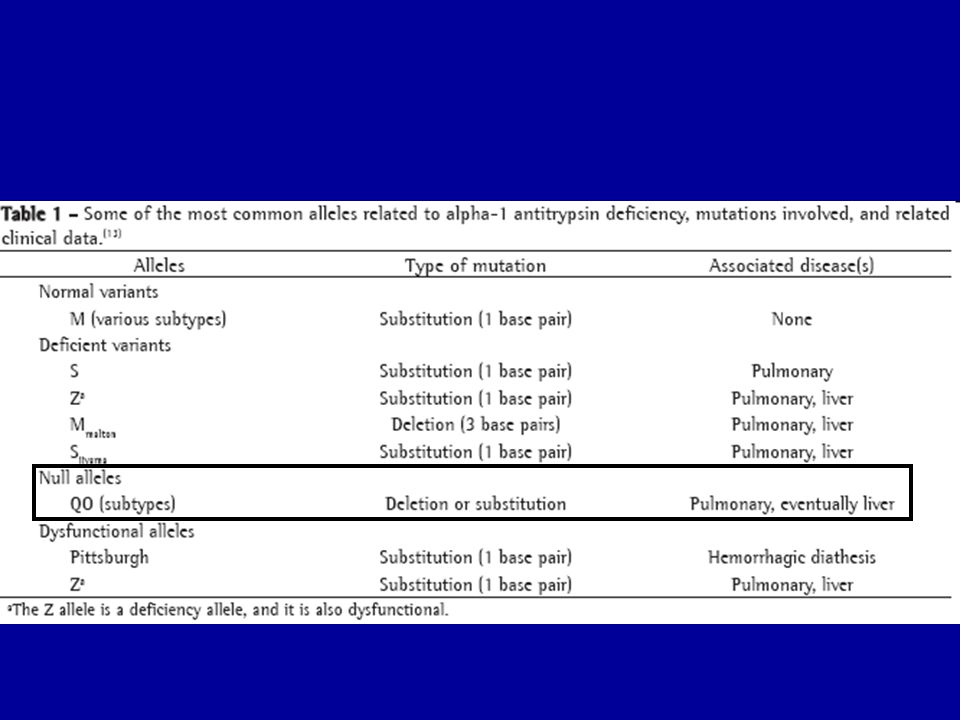
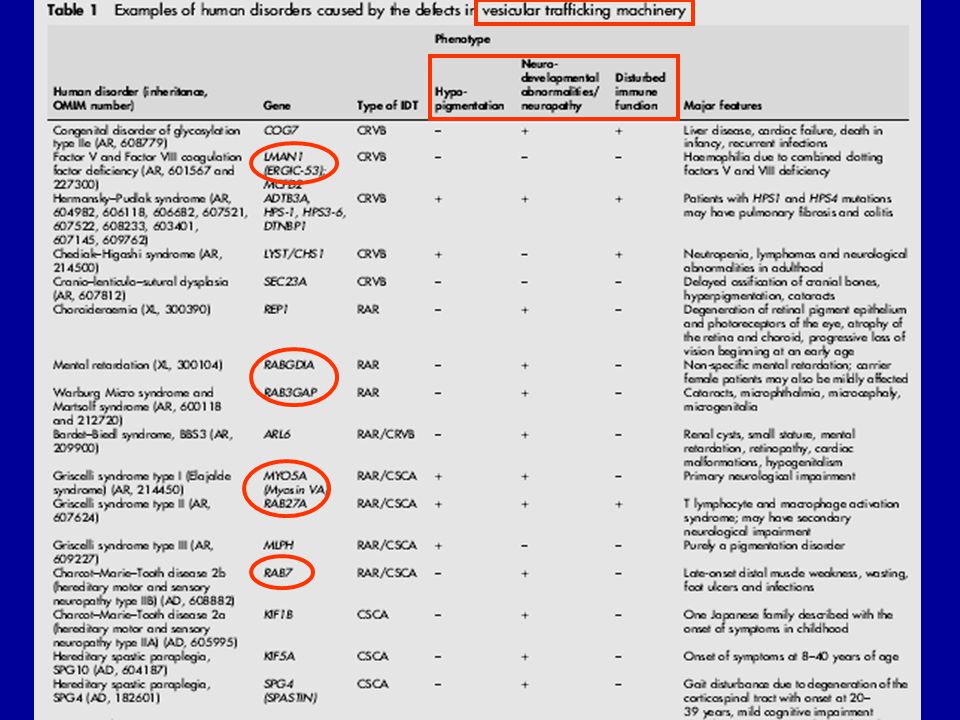
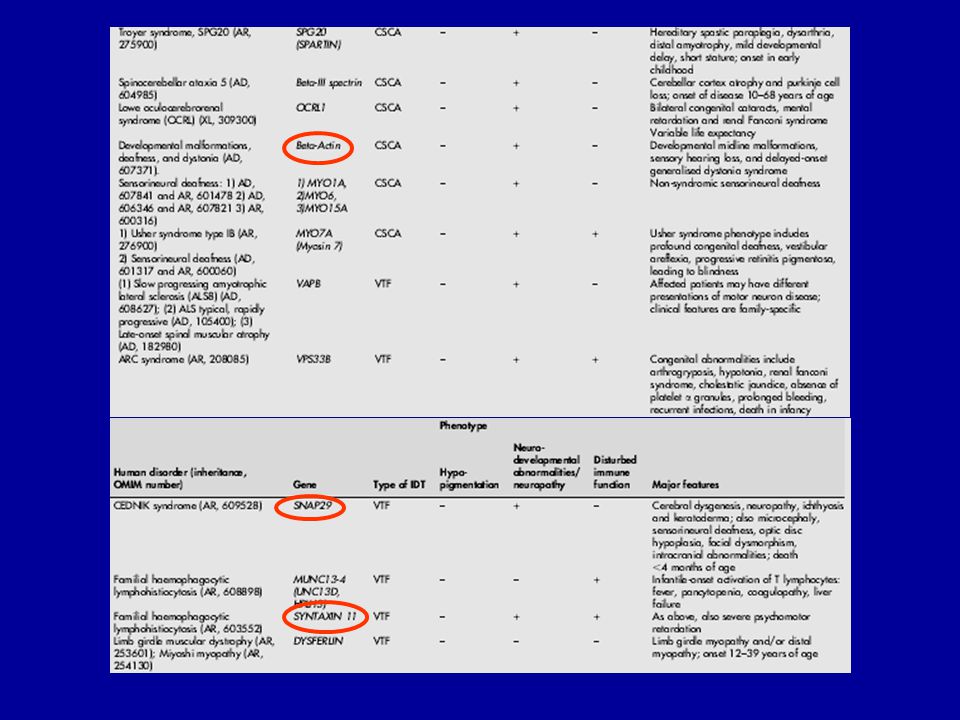
Protein hiány (ER retenció) Cystic fibrosis and associated diseases a1-antitrypsin deficiency without liver disease Congenital hypothyroidism: Thyroglobulin deficiency Thyroid peroxidase deficiency Thyroxin binding globulin deficiency Protein C deficiency Disorders of lipid metabolism LDL receptor defect Lipoprotein lipase deficiency Lipoprotein(a) deficiency Hereditary hypoparathyroidism Nephrogenic diabetes insipidus due to mutations in AVP receptor 2 or aquaporin-2 Growth hormone receptor deficiency Osteogenesis imperfecta Procollagen type I, II, IV deficiency Albinism/tyrosinase deficiency Obesity/elevated prohormone levels: prohormone convertase 1 deficiency 2. Toxikus protein vagy protein aggregátumok Autosomal dominant neurohypophyseal diabetes insipidus (aquaporin-2) Liver disease in a1-antitrypsin deficiency ? Creutzfeldt-Jakob disease ? Retinitis pigmentosa 3. Hibás transzport mechanizmus Combined coagulation factor V and VIII deficiency (ERGIC-53 mutation) Abetalipoproteinemia/deficiency of microsomal triglyceride transfer protein .
 Cystic fibrosis and associated diseases. a1-antitrypsin deficiency without liver disease. Congenital hypothyroidism: Thyroglobulin deficiency. Thyroid peroxidase deficiency. Thyroxin binding globulin deficiency. Protein C deficiency. Disorders of lipid metabolism. LDL receptor defect. Lipoprotein lipase deficiency. Lipoprotein(a) deficiency. Hereditary hypoparathyroidism. Nephrogenic diabetes insipidus due to mutations in AVP receptor 2 or aquaporin-2. Growth hormone receptor deficiency. Osteogenesis imperfecta. Procollagen type I, II, IV deficiency. Albinism/tyrosinase deficiency. Obesity/elevated prohormone levels: prohormone convertase 1 deficiency. 2. Toxikus protein vagy protein aggregátumok. Autosomal dominant neurohypophyseal diabetes insipidus (aquaporin-2) Liver disease in a1-antitrypsin deficiency. Creutzfeldt-Jakob disease. Retinitis pigmentosa. 3. Hibás transzport mechanizmus. Combined coagulation factor V and VIII deficiency (ERGIC-53 mutation) Abetalipoproteinemia/deficiency of microsomal triglyceride transfer protein. .")
13
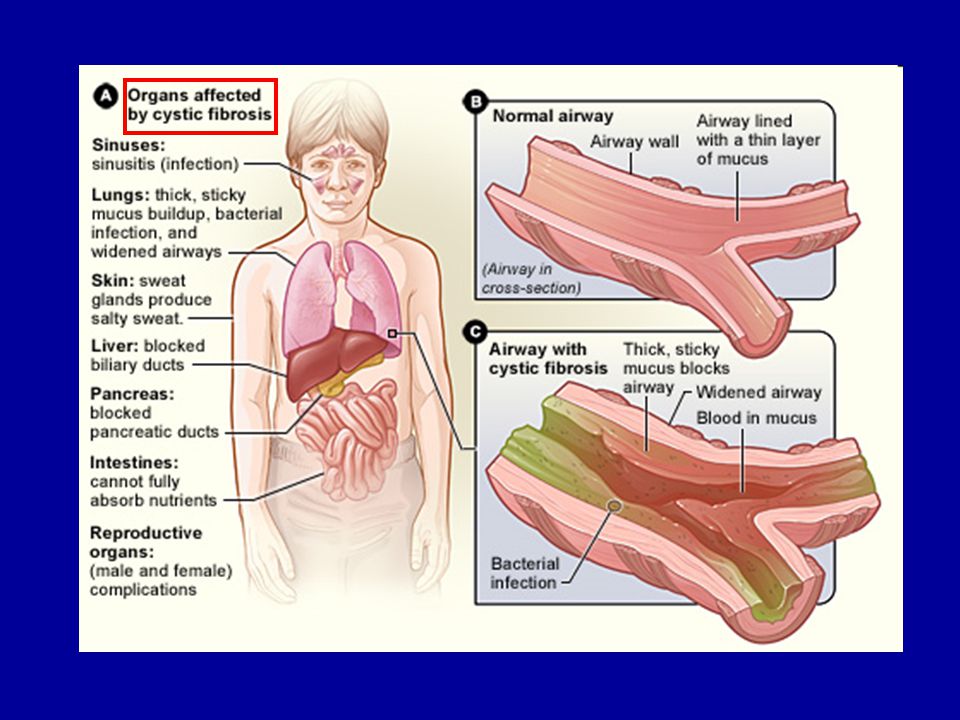
Cisztás fibrózis (AR) Nincs Cl- és víz transzport, sűrű nyálkaréteg,
Fertőzések (tüdő)
")
14
Egy génnek különböző mutációi
Alléljei vannak (multiplex allélizmus)
")
15
CFTR szerkezete NBF = ATP kötőhely F508 – 508. aminosav hiányzik

16
CFTR transzport F508 CFTR fehérje hajtogatódása nem jó,
lebomlik a fehérje, nincs CFTR a plazmamembránban Transport of CFTR through the cell. Southern K W Arch Dis Child 1997;76: ©1997 by BMJ Publishing Group Ltd and Royal College of Paediatrics and Child Health

18
Cisztás fibrózis génterápiája

19
Protein hiány(nincs transzport az ER-ből)
Endoplazmás retikulum tárolási betegségek Protein hiány (ER retenció) Cystic fibrosis and associated diseases a1-antitrypsin deficiency without liver disease Congenital hypothyroidism: Thyroglobulin deficiency Thyroid peroxidase deficiency Thyroxin binding globulin deficiency Protein C deficiency Disorders of lipid metabolism LDL receptor defect Lipoprotein lipase deficiency Lipoprotein(a) deficiency Hereditary hypoparathyroidism Nephrogenic diabetes insipidus due to mutations in AVP receptor 2 or aquaporin-2 Growth hormone receptor deficiency Osteogenesis imperfecta Procollagen type I, II, IV deficiency Albinism/tyrosinase deficiency Obesity/elevated prohormone levels: prohormone convertase 1 deficiency 2. Toxikus protein vagy protein aggregátumok Autosomal dominant neurohypophyseal diabetes insipidus (aquaporin-2) Liver disease in a1-antitrypsin deficiency ? Creutzfeldt-Jakob disease ? Retinitis pigmentosa 3. Hibás transzport mechanizmus Combined coagulation factor V and VIII deficiency (ERGIC-53 mutation) Abetalipoproteinemia/deficiency of microsomal triglyceride transfer protein . Protein hiány(nincs transzport az ER-ből)
 Cystic fibrosis and associated diseases. a1-antitrypsin deficiency without liver disease. Congenital hypothyroidism: Thyroglobulin deficiency. Thyroid peroxidase deficiency. Thyroxin binding globulin deficiency. Protein C deficiency. Disorders of lipid metabolism. LDL receptor defect. Lipoprotein lipase deficiency. Lipoprotein(a) deficiency. Hereditary hypoparathyroidism. Nephrogenic diabetes insipidus due to mutations in AVP receptor 2 or aquaporin-2. Growth hormone receptor deficiency. Osteogenesis imperfecta. Procollagen type I, II, IV deficiency. Albinism/tyrosinase deficiency. Obesity/elevated prohormone levels: prohormone convertase 1 deficiency. 2. Toxikus protein vagy protein aggregátumok. Autosomal dominant neurohypophyseal diabetes insipidus (aquaporin-2) Liver disease in a1-antitrypsin deficiency. Creutzfeldt-Jakob disease. Retinitis pigmentosa. 3. Hibás transzport mechanizmus. Combined coagulation factor V and VIII deficiency (ERGIC-53 mutation) Abetalipoproteinemia/deficiency of microsomal triglyceride transfer protein. . Protein hiány(nincs transzport az ER-ből)")
20
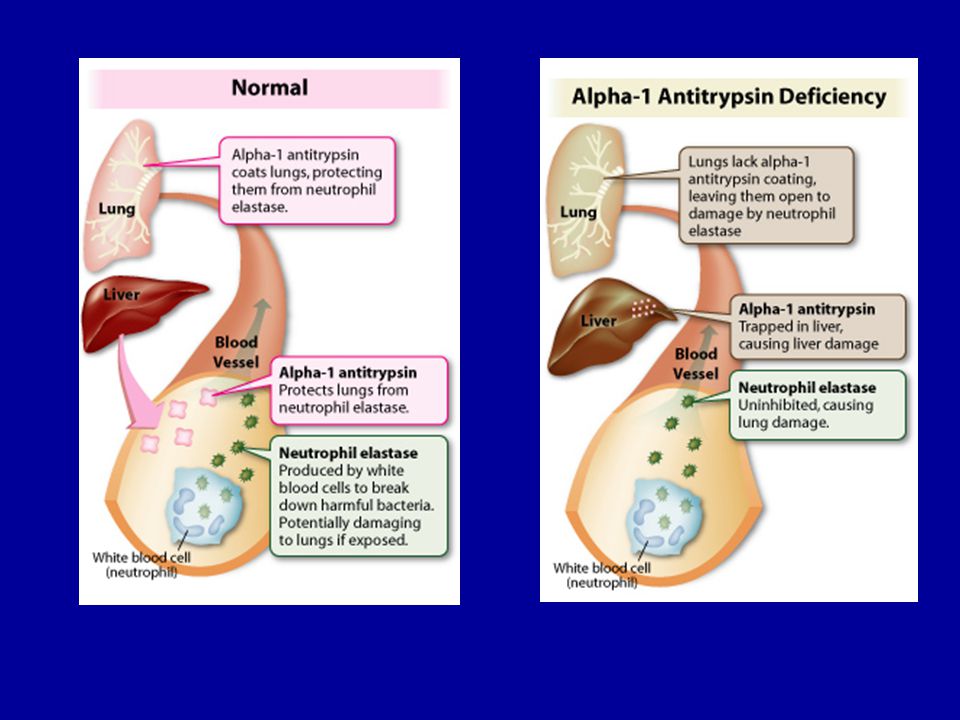
Alfa1 antitripszin funkciója
Proteáz inhibitor, a máj termeli Fertőzések neutrofil granulociták elasztázt (proteáz) bocsájtanak ki elpusztítják a kórokozókat, de ha nincs megfelelő mennyiségben alfa1 antitripszin, a tüdő szövetei is roncsolódnak (az elasztáz és az alfa1 antitripszin egyensúlya szükséges) emphysema (csökken a légzőfelület) Homozigótákban expresszálódik
 bocsájtanak ki elpusztítják a kórokozókat, de ha. nincs megfelelő mennyiségben alfa1 antitripszin, a tüdő szövetei is roncsolódnak (az elasztáz és az alfa1 antitripszin. egyensúlya szükséges) emphysema (csökken a légzőfelület) Homozigótákban expresszálódik.")
22
Heterozigótákban a dohányzás
kiválthatja Dohányzás – több elasztáz

23
Alfa1 antitripszin hiány oka és következményei
Génmutáció következményeként rosszul hajtogatott fehérje keletkezik, ami az ER minőség ellenőrző működése következtében lebomlik – hiányzik a fehérje emphysema alakul ki A betegek kisebb százalékában májkárosodást is okoz, nehezen vagy egyáltalán nem bomlik le a fehérje (aggregátumok jönnek létre, amelyek gátolják a sejtek működését)
")
25
Alpha1 antitripszin akkumuláció hepatocitákban
HE immunohisztokémia

27
Endoplazmás retikulum tárolási betegségek
Protein hiány (ER retenció) Cystic fibrosis and associated diseases a1-antitrypsin deficiency without liver disease Congenital hypothyroidism: Thyroglobulin deficiency Thyroid peroxidase deficiency Thyroxin binding globulin deficiency Protein C deficiency Disorders of lipid metabolism LDL receptor defect Lipoprotein lipase deficiency Lipoprotein(a) deficiency Hereditary hypoparathyroidism Nephrogenic diabetes insipidus due to mutations in AVP receptor 2 or aquaporin-2 Growth hormone receptor deficiency Osteogenesis imperfecta Procollagen type I, II, IV deficiency Albinism/tyrosinase deficiency Obesity/elevated prohormone levels: prohormone convertase 1 deficiency 2. Toxikus protein vagy protein aggregátumok Autosomal dominant neurohypophyseal diabetes insipidus (aquaporin-2) Liver disease in a1-antitrypsin deficiency ? Creutzfeldt-Jakob disease ? Retinitis pigmentosa 3. Hibás transzport mechanizmus Combined coagulation factor V and VIII deficiency (ERGIC-53 mutation) Abetalipoproteinemia/deficiency of microsomal triglyceride transfer protein .
 Cystic fibrosis and associated diseases. a1-antitrypsin deficiency without liver disease. Congenital hypothyroidism: Thyroglobulin deficiency. Thyroid peroxidase deficiency. Thyroxin binding globulin deficiency. Protein C deficiency. Disorders of lipid metabolism. LDL receptor defect. Lipoprotein lipase deficiency. Lipoprotein(a) deficiency. Hereditary hypoparathyroidism. Nephrogenic diabetes insipidus due to mutations in AVP receptor 2 or aquaporin-2. Growth hormone receptor deficiency. Osteogenesis imperfecta. Procollagen type I, II, IV deficiency. Albinism/tyrosinase deficiency. Obesity/elevated prohormone levels: prohormone convertase 1 deficiency. 2. Toxikus protein vagy protein aggregátumok. Autosomal dominant neurohypophyseal diabetes insipidus (aquaporin-2) Liver disease in a1-antitrypsin deficiency. Creutzfeldt-Jakob disease. Retinitis pigmentosa. 3. Hibás transzport mechanizmus. Combined coagulation factor V and VIII deficiency (ERGIC-53 mutation) Abetalipoproteinemia/deficiency of microsomal triglyceride transfer protein. .")
28
A pajzsmirigy jódtartalmú hormonjainak termelése
follikulusok colloid Jodid felvétel, átszállítás és leadás a lumenbe Tireoglobulin (sok tirozin) szintézis Peroxidáz (TPO) szintézis és leadás peroxidáz jódozza a tireoglobulin tirozinjait (MIT, DIT) tárolás
 szintézis. Peroxidáz (TPO) szintézis és leadás. peroxidáz jódozza a tireoglobulin tirozinjait (MIT, DIT) tárolás.")
29
A pajzsmirigy jódtartalmú hormonjainak termelése
follikulusok Tireoglobulin felvétele, emésztése és a T3 (trijódtironin) és T4 (tiroxin) felszabadul T3 és T4 leadása a vérbe a vérben tiroxin kötő fehérjéhez kötődik (TBG)
 és T4 (tiroxin) felszabadul. T3 és T4 leadása a vérbe. a vérben tiroxin kötő fehérjéhez kötődik (TBG)")
30
A pajzsmirigy jódtartalmú hormonjainak termelése

31
Veleszületett hypothyroidismus okai
A hormonok szintézisében ill. működésében szerepet játszó fehérjék hiánya: pl. a mutáns fehérje rosszul hajtogatódik (ER-ben marad), nem kerül ki a sejtből Tireoglobulin hiány Pajzsmirigy (thyroid) peroxidáz (TPO) hiány Tiroxin kötő fehérje hiány
, nem kerül ki a sejtből. Tireoglobulin hiány. Pajzsmirigy (thyroid) peroxidáz (TPO) hiány. Tiroxin kötő fehérje hiány.")
32
Endoplazmás retikulum tárolási betegségek
Protein hiány (ER retenció) Cystic fibrosis and associated diseases a1-antitrypsin deficiency without liver disease Congenital hypothyroidism: Thyroglobulin deficiency Thyroid peroxidase deficiency Thyroxin binding globulin deficiency Protein C deficiency Disorders of lipid metabolism LDL receptor defect Lipoprotein lipase deficiency Lipoprotein(a) deficiency Hereditary hypoparathyroidism Nephrogenic diabetes insipidus due to mutations in AVP receptor 2 or aquaporin-2 Growth hormone receptor deficiency Osteogenesis imperfecta Procollagen type I, II, IV deficiency Albinism/tyrosinase deficiency Obesity/elevated prohormone levels: prohormone convertase 1 deficiency 2. Toxikus protein vagy protein aggregátumok Autosomal dominant neurohypophyseal diabetes insipidus (aquaporin-2) Liver disease in a1-antitrypsin deficiency ? Creutzfeldt-Jakob disease ? Retinitis pigmentosa 3. Hibás transzport mechanizmus Combined coagulation factor V and VIII deficiency (ERGIC-53 mutation) Abetalipoproteinemia/deficiency of microsomal triglyceride transfer protein .
 Cystic fibrosis and associated diseases. a1-antitrypsin deficiency without liver disease. Congenital hypothyroidism: Thyroglobulin deficiency. Thyroid peroxidase deficiency. Thyroxin binding globulin deficiency. Protein C deficiency. Disorders of lipid metabolism. LDL receptor defect. Lipoprotein lipase deficiency. Lipoprotein(a) deficiency. Hereditary hypoparathyroidism. Nephrogenic diabetes insipidus due to mutations in AVP receptor 2 or aquaporin-2. Growth hormone receptor deficiency. Osteogenesis imperfecta. Procollagen type I, II, IV deficiency. Albinism/tyrosinase deficiency. Obesity/elevated prohormone levels: prohormone convertase 1 deficiency. 2. Toxikus protein vagy protein aggregátumok. Autosomal dominant neurohypophyseal diabetes insipidus (aquaporin-2) Liver disease in a1-antitrypsin deficiency. Creutzfeldt-Jakob disease. Retinitis pigmentosa. 3. Hibás transzport mechanizmus. Combined coagulation factor V and VIII deficiency (ERGIC-53 mutation) Abetalipoproteinemia/deficiency of microsomal triglyceride transfer protein. .")
33
Vese eredetű diabetes insipidus
nem megfelelő a vesében a víz visszaszívása ok: - nem működik a vazopresszin receptor - aquaporin nem működik A víz diffúzióval és fehérje (aquaporin) segítségével mehet át a membránon.
 segítségével mehet át a membránon.")
34
In the case of given mutation of AVP receptor OR aquaporin causing
Blood Collecting Duct lumen Under normal physiological conditions, decreases in blood pressure or blood volume and increases in extracellular sodium cc. Arginine vasopressin (AVP) is released into circulation, causing water retention in the renal collecting duct cell. In the collecting duct cell, AVP targets the V2 receptor, activating protein kinase by way of cAMP and phosphorylates aquaporins that insert into the luminal cell membrane of the collecting duct.With aquaporins in place, large volumes of water are reabsorbed from the tubular fluid, subsequently increasing body water, blood pressure, and blood volume. In the case of given mutation of AVP receptor OR aquaporin causing misfolding of the proteins in ER, the proteins are degraded and not transported to plasmamembrane.
 is released into. circulation, causing water retention in the. renal collecting duct cell. In the collecting duct cell, AVP targets. the V2 receptor, activating protein kinase. by way of cAMP and phosphorylates aquaporins. that insert into the luminal cell membrane of. the collecting duct.With aquaporins in place, large volumes of water are reabsorbed from. the tubular fluid, subsequently increasing. body water, blood pressure, and blood volume. In the case of given mutation of. AVP receptor OR aquaporin causing. misfolding of the proteins in ER, the. proteins are degraded and not. transported to plasmamembrane.")
35
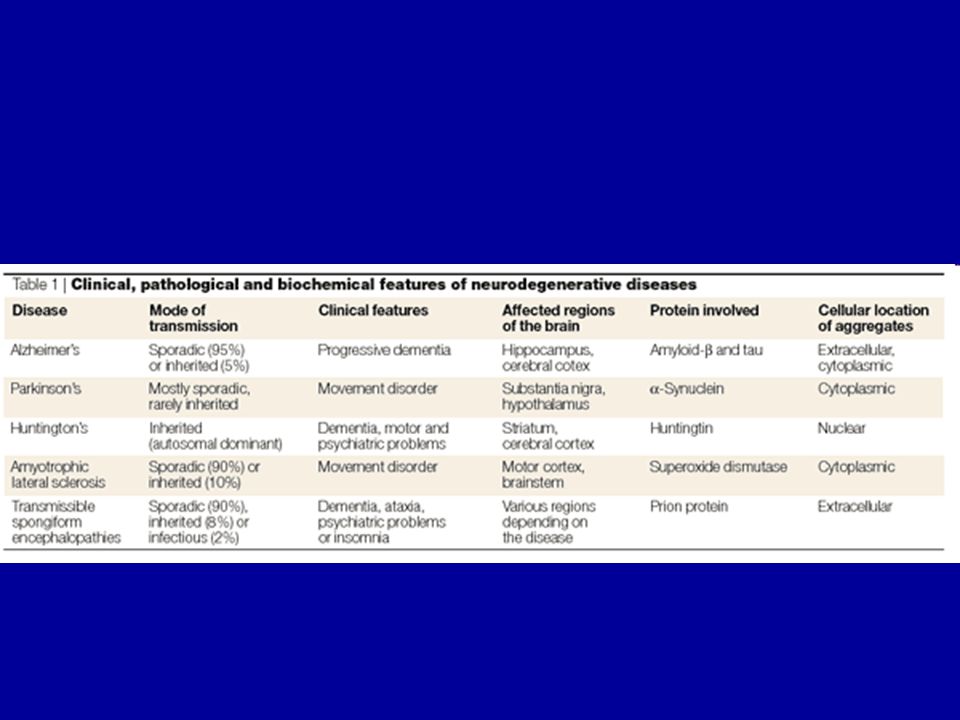
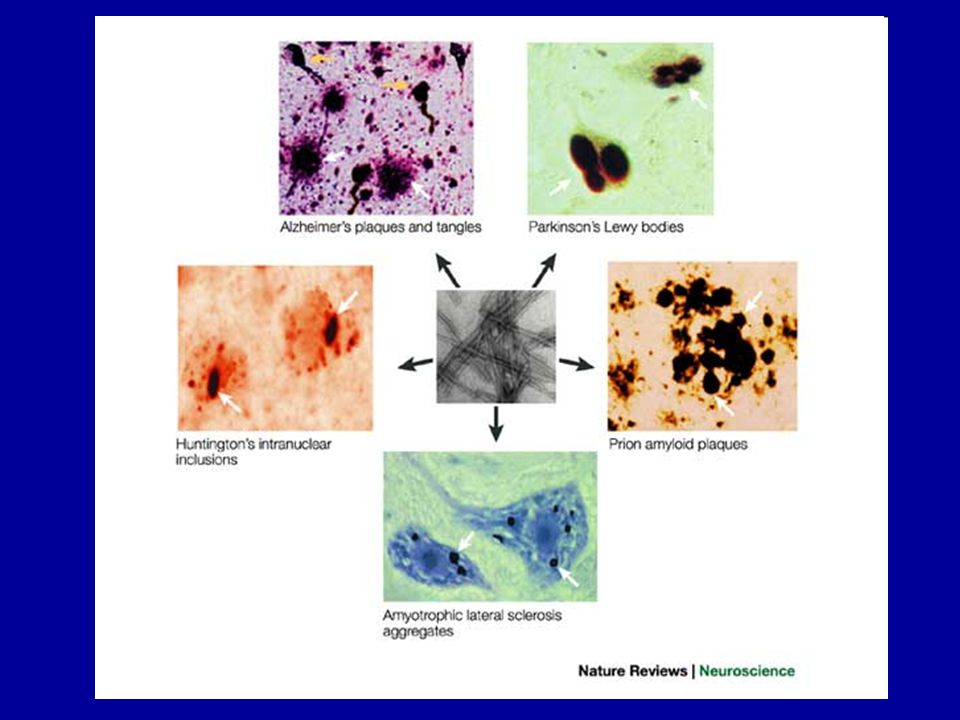
Rosszul hajtogatott fehérjék szerepe a
neurodegeneratív betegségekben

37
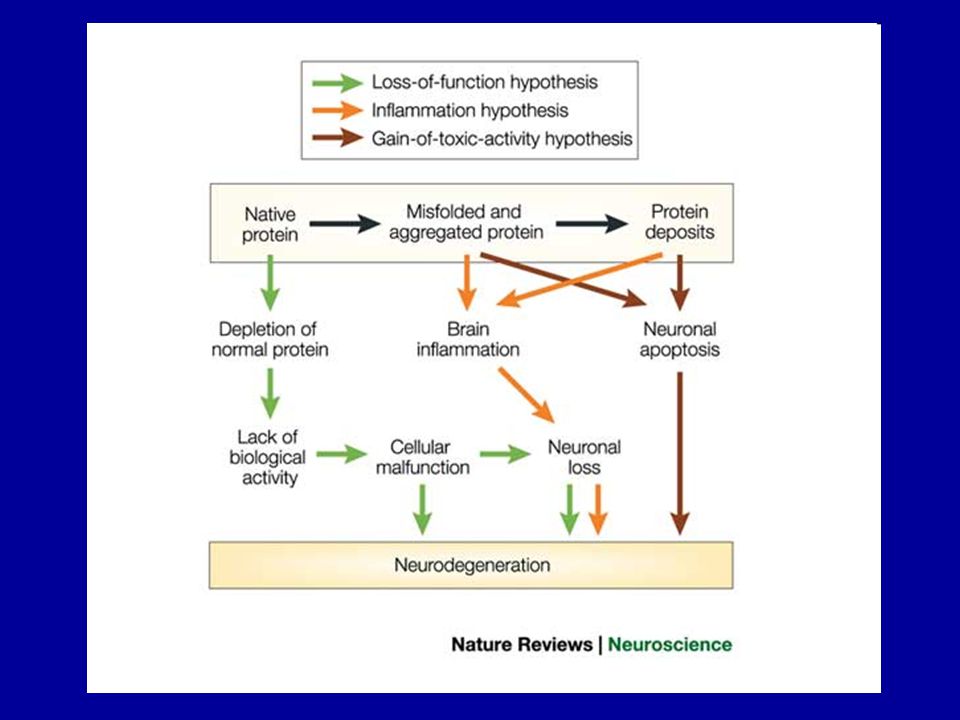
Summary of proposed involvement of protein misfolding in neurodegenerative diseases

41
Az eukarióta sejt legjelentősebb transzportja a vezikuláris transzport

42
Vezikuláris transzport
mechanizmusa

43
Vezikuláris transzport főbb lépései
Fehérjék kiválogatása, majd vezikulumba csomagolása ligand – receptor – burok képző fehérje (+adaptor) kis G protein
 kis G protein.")
44
A vezikulumok tartalma specifikus
citoszol Specifikus kapcsolódás Burok fehérje Adaptor fehérje lumen Receptorok és burkok biztosítják

45
A burok fehérjék (kívül)
Klatrin COPI és II kaveolin A vezikulum burkai előbb (klatrin) vagy utóbb (COP) leválnak. leválás kialakulás energiaigényes
 vagy utóbb (COP) leválnak. leválás kialakulás energiaigényes.")
46
A burkok útvonal specifikusak

47
Klatrin szerkezete és a
klatrin burkos vezikulum

48
Vezikuláris transzport főbb lépései
Fehérjék kiválogatása, majd vezikulumba csomagolása ligand – receptor – burok képző fehérje (adaptor) kis G protein 2. Vezikulum lefűződése speciális foszfolipid összetétel (a membránban) mechanoenzim (a vezikulum lefűzéséhez)
 kis G protein. 2. Vezikulum lefűződése. speciális foszfolipid összetétel (a membránban) mechanoenzim (a vezikulum lefűzéséhez)")
49
A klatrin burkos vezikulumok lefűződése egy
mechanoenzim, a dinamin (monomer G protein) segítségével történik
 segítségével történik.")
50
Vezikuláris transzport főbb lépései
Fehérjék kiválogatása, majd vezikulumba csomagolása ligand – receptor – burok képző fehérje (adaptor) kis G protein 2. Vezikulum lefűződése speciális foszfolipid összetétel (a membránban) mechanoenzim (a vezikulum lefűzéséhez) 3. A vezikulum szállítása diffúzió, citoszkeleton
 kis G protein. 2. Vezikulum lefűződése. speciális foszfolipid összetétel (a membránban) mechanoenzim (a vezikulum lefűzéséhez) 3. A vezikulum szállítása. diffúzió, citoszkeleton.")
51
A vezikulumok a citoszkeleton segítségével szállítódnak
Aktin filamentum (miozin) mikrotubulus (dinein vagy kinezin)
 mikrotubulus. (dinein vagy. kinezin)")
52
Vezikuláris transzport főbb lépései
Fehérjék kiválogatása, majd vezikulumba csomagolása ligand – receptor – burok képző fehérje (adaptor) kis G protein 2. Vezikulum lefűződése speciális foszfolipid összetétel (a membránban) mechanoenzim (a vezikulum lefűzéséhez) 3. A vezikulum szállítása diffúzió, citoszkeleton 4. Célmembrán megtalálása és dokkolás komplemeter v és tSNARE-k Rab (monomer G) fehérjék
 kis G protein. 2. Vezikulum lefűződése. speciális foszfolipid összetétel (a membránban) mechanoenzim (a vezikulum lefűzéséhez) 3. A vezikulum szállítása. diffúzió, citoszkeleton. 4. Célmembrán megtalálása és dokkolás. komplemeter v és tSNARE-k. Rab (monomer G) fehérjék.")
53
Hogyan találják meg a vezikulumok a célmembránjukat?
Donor kompartment Célkompartment 1 Dokkolás fúzió Célkompartment 2

54
Monomer (kis) G-fehérjék
 G-fehérjék")
55
GTP kötött formában beépül a membránba GDP kötött formában szolubilis
Rab monomer G fehérje szerepe a dokkolásban Rab fehérje: GTP kötött formában beépül a membránba GDP kötött formában szolubilis

56
A Rab monomer G proteinek
specifikusak az adott vezikuláris transzportra

57
Vezikuláris transzport főbb lépései
Fehérjék kiválogatása, majd vezikulumba csomagolása ligand – receptor – burok képző fehérje (adaptor) kis G protein 2. Vezikulum lefűződése speciális foszfolipid összetétel (a membránban) mechanoenzim (a vezikulum lefűzéséhez) 3. A vezikulum szállítása diffúzió, citoszkeleton 4. Célmembrán megtalálása és dokkolás komplemeter v és tSNARE-k Rab (monomer G) fehérjék 5. Fúzió SNARE-ek és fúziós fehérjék SNARE-ek szétválasztása (NSF)
 kis G protein. 2. Vezikulum lefűződése. speciális foszfolipid összetétel (a membránban) mechanoenzim (a vezikulum lefűzéséhez) 3. A vezikulum szállítása. diffúzió, citoszkeleton. 4. Célmembrán megtalálása és dokkolás. komplemeter v és tSNARE-k. Rab (monomer G) fehérjék. 5. Fúzió SNARE-ek és fúziós fehérjék. SNARE-ek szétválasztása (NSF)")
58
Membrán fúzió Nature Medicine 8, (2002)
")
59
Kapcsolódó SNARE-eket az NSF választja szét

60
Neurotoxinok amelyek az idegsejtek vezikuláris transzportját (SNARE-ket) gátolják
Tetanus toxin (Clostridium tetani) gátló neurotranszmitterek nem tudnak kiürülni (nincs exocitózis) Botulinum toxin (Clostridium botulinum) stimuláló neurotranszmitterek nem tudnak kiürülni
 gátló neurotranszmitterek nem. tudnak kiürülni (nincs exocitózis) Botulinum toxin. (Clostridium botulinum) stimuláló neurotranszmitterek nem. tudnak kiürülni.")
63
Az eukarióta sejt legjelentősebb transzportja a vezikuláris transzport

65
A szekréciós útvonal Golgi

66
A Golgi készüléket Camillo Golgi fedezte fel
Internal reticular apparatus (1897) – Golgi komplex – Golgi Nobel díj 1906 Fém impregnáció
 – Golgi komplex – Golgi. Nobel díj Fém impregnáció.")
67
A Golgi szerkezete polarizált (szerkezet és funkció)
Fénymikroszkópos kép (immuncitokémia) Transzmissziós elektron mikroszkópos kép Sémás kép
 Transzmissziós elektron. mikroszkópos kép. Sémás kép.")
68
A Golgi alkompartmentjei

69
Golgi ER-Golgi intermedier kompartment (ERGIC-53)
Endoplazmás retikulum

70
A véralvadás sémája Kombinált hiányuk oka, az ERGIC-53 fehérje
X-kromoszómához kötött Kombinált hiányuk oka, az ERGIC-53 fehérje mutációja (csaperon fehérje, amely a VIII és V faktor szekréciójához kell) A faktorok a májban termelődnek
 A faktorok a májban termelődnek.")
71
Mikrotubulusok szerepe a Golgi szerkezet fenntartásában
2002 by Bruce Alberts, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter. Intakt MT depolimerizált MT Golgi- zöld Mikrotubulus (MT) - piros
 - piros.")
72
Szénhidrátlánc átalakítás Szénhidrátlánc szintézis
Mi történik a Golgiban? dER ERGIC Cisz Golgi hálózat (CGN) Cisz Golgi Mediális Golgi Transz Golgi Transz Golgi hálózat (TGN) Szénhidrátlánc átalakítás Szénhidrátlánc szintézis Osztályozás
 Cisz Golgi. Mediális Golgi. Transz Golgi. Transz Golgi hálózat. (TGN) Szénhidrátlánc átalakítás. Szénhidrátlánc szintézis. Osztályozás.")
73
Golgi funkciói N- oligoszacharid lánc módosítása, új oligo- vagy
poliszacharid láncok kötése - M-6-PO4 csoport (lizoszómális enzimek) - nincs változtatás (sok mannóz) - mannózok cseréje más monoszacharidokra - O-glikoziláció - proteoglikán szintézis SO4 csoport kötése aminosavakra vagy szénhidrátláncra lipidek módosítása – glikolipidek, - szfingomielin proteolízis osztályozás – vissza az ER-be - Golgi proteinek visszatartása - TGN – lizoszóma, plazmamembrán
 - nincs változtatás (sok mannóz) - mannózok cseréje más monoszacharidokra. - O-glikoziláció. - proteoglikán szintézis. SO4 csoport kötése aminosavakra vagy szénhidrátláncra. lipidek módosítása – glikolipidek, - szfingomielin. proteolízis. osztályozás – vissza az ER-be. - Golgi proteinek visszatartása. - TGN – lizoszóma, plazmamembrán.")
74
Glikoziláció Oligoszacharid lánc kapcsolása fehérjéhez (lipidhez)
Glikozil transzferázok katalizálják N-glikoziláció (Asn) ER-ben történik, ER-ben és Golgiban módosul (hosszabb lánc) O-glikoziláció (Ser, Thr) Golgiban történik (rövidebb láncok)
 ER-ben történik, ER-ben és Golgiban módosul. (hosszabb lánc) O-glikoziláció (Ser, Thr) Golgiban történik (rövidebb láncok)")
75
Glikoziláció

76
Proteoglikánok Plazmamembrán ECM
N and O oligosaccharides are not shown A fehérjéhez ismétlődő diszacharidokból (negatív töltésű - COOH, -SO4) álló glükózaminoglikánok (pl. hialuronsav, kondroitin szulfát, dermatán szulfát és heparán szulfát) poliszacharidok kapcsolódnak.
 álló glükózaminoglikánok (pl. hialuronsav, kondroitin szulfát, dermatán szulfát és heparán szulfát) poliszacharidok kapcsolódnak.")
77
Membránlipid szintézis

78
Kétirányú transzport a dER és a Golgi között
anterográd retrográd ER retenciós szignál KDEL a C terminális végen

79
A plazmamembrán felé irányuló transzport:
konstitutív és a regulált szekréció (exocitózis) Minden sejt állandóan pl. membrán pótlás, ECM szignál Csak bizonyos sejtek, ingerre idegsejtek és mirigy sejtek
 Minden sejt állandóan. pl. membrán pótlás, ECM. szignál. Csak bizonyos sejtek, ingerre. idegsejtek és mirigy. sejtek.")
80
Polarizált sejtben a konstitutív szekrécióval
a megfelelő helyre kell eljuttatni a membránkomponenseket

82
Néhány genetikai betegség, amely hibás Golgi működésre vezethető vissza

83
Cu metabolizmus Bélhámsejt májsejt ATP7B ATP7A Vér

84
Menkes kór - ATP7A mutáció – XR
– Cu hiány - enzimek működéséhez kell Wilson kór - ATP7B (máj,idegrendszer) XR – Cu felhalmozódás
 XR. – Cu felhalmozódás.")
85
Fehérjék, amelyek hibája izomdisztrófiát okoz

86
Glikoziláció hiányában nincs kapcsolódás
A fehérjék glikozilációja a Golgiban történik.

87
Golgi glikozilációs enzimek, amelyek hibája izomdisztrófiát okoz

88
I (inclusion) sejt betegség
Golgi - foszfotranszferáz hiány – lizoszómális enzimek nem kapnak M-6-P szignált, ezért szekrécióra kerülnek, a sejtbe került anyagok nem tudnak lebomlani =inclusion body-k Főleg fibroblaszt és makrofág ( más sejtekben más útvonal lehet?) letális
 letális.")





: Tiroxin T4>")



 prokariótában>")












