Előadást letölteni
1
A haemostasis és betegségeinek genetikai vonatkozásai

2
ÖRÖKLETES RENDELLENESSÉGEK
Thrombofíliák Koagulopátiák A thrombocyták veleszületett rendellenességei

3
THROMBOPHILIA: FOKOZOTT THROMBOSIS HAJLAMMAL JÁRÓ ÁLLAPOTOK
Veleszületett Szerzett

4
TROMBOFÍLIÁRA HÍVJA FEL A FIGYELMET
Fiatal korban (50) kialakuló trombózis Szokatlan helyen kialakuló trombózis Recidíváló trombózis Enyhe provokáló tényező mellett kialakuló trombózis Habituális vetélés, koraszülés, halvaszülés
 kialakuló trombózis. Szokatlan helyen kialakuló trombózis. Recidíváló trombózis. Enyhe provokáló tényező mellett kialakuló trombózis. Habituális vetélés, koraszülés, halvaszülés.")
5
ÖRÖKLETES TROMBOFÍLIÁK
genetikai hiba Családi halmozódás Általában vénás trombózisok gyakorisága nő meg Gyakori eltérések enyhe trombózis hajlammal járnak Ritkábbak súlyosabbal

6
GYAKORIBB ÖRÖKLETES TROMBOFÍLIÁK
VF Leiden mutáció Aktivált protein C rezisztencia Prothrombin gén mutáció Hyperhomocysteinaemia Emelkedett FVIII szint

7
RITKÁBB ÖRÖKLETES TROMBOFÍLIÁK
Antithrombin III defektus Protein C rendszer rendellenessége Protein S defektus VF Leiden mutáció - homozygota Prothrombin gén mutáció - homozygota

8
TÖRTÉNETI ÁTTEKINTÉS Egeberg, AT defektus első familiáris trombofília leírása protein C defektus Magyarországon az első protein C hiányos család, Domján protein S hiányos család APC rezisztencia, Dahlback 1994 FV Leiden mutáció, Bertina

9
ÖRÖKLETES THROMBOPHILIÁK
Antithrombin III defektus Protein C rendszer rendellenessége Protein S defektus Aktivált protein C rezisztencia VF Leiden mutáció Prothrombin gén mutáció Hyperhomocysteinaemia „Sticky platelet syndrome” Dysfibrinogenaemia Thrombomodulin mutációk Plasminogén defektus Heparin kofaktor II hiány Szöveti faktor út gátló (TFPI) hiány XII F részleges hiánya Genetikusan determinált emelkedett VIII F szinttel járó állapot Az ok általában %-ban tisztázható
 hiány. XII F részleges hiánya. Genetikusan determinált emelkedett VIII F szinttel járó állapot. Az ok általában %-ban tisztázható.")
10
TERMÉSZETES ANTIKOAGULÁNSOK
Az alvadási folyamat inhibitorai Szerep: a trombus képződés az érsérülés helyén lokalizált maradjon

11
Antithrombin III – heparin rendszer
AT III szerepe: serin proteáz inhibitor Aktivált alvadási faktorok irreverzibilis inaktiválása Xa / IIa / Ixa / XIa inaktivációjában vesz részt Hatását a heparin, és az EC felszínén lévő heparán szulfát fokozza

12
Antithrombin III – heparin rendszer
Fontos: AT hiányában a heparin kevésbé hat!

13
AT – heparin – trombin komplex

14
Antithrombin III defektus
Az első felismert örökletes thrombophilia Egeberg 1965, első ATIII hiányos család Általában nem teljes hiány, csak kb 50 %-os csökkenés Hiány, csökkenés, funkcionális eltérések Mutációk heterogén csoportja, okozhat funkció csökkenést is Lehet szerzett is (pl. májbetegségekben) Autoszomális domináns öröklődés kódoló gén az 1. kromoszómán van
 Autoszomális domináns öröklődés. kódoló gén az 1. kromoszómán van.")
15
Antithrombin III defektus
Felosztás: I. mennyiségi csökkenés homozygota nem életképes, iu. meghal heterozygota: 50 %-os ag csökkenés 20 %-os trombózis rizikó Már fiatal korban MVT, OAK szedés mellett már az első 1-2 hónap során MVT

16
Antithrombin III defektus
II. Funkcionális eltérés, normális ag szint antigén szint normális, vagy enyhén csökkent, kóros működés, heterogén csoport: attól függően, hogy hol van a mutáció reaktív, vagy heparin-kötő receptort érinti Reaktív kötőhely mutációi: trombint nem képest közömbösíteni ritka, 20 x rizikó Heparin kötőhely mutációi: ritka homozygota mutáció: már gyermekkorban súlyos MVT heterozygota: gyakran tünetmentes

17
Antithrombin III defektus
20x thrombosis rizikó Más genetikai zavarral társulva nagyobb rizikó Ritka, MVT 1%-a, I. típus: egészséges populáció 0,02% I-II. típus: 0,16 %

19
A PROTEIN C RENDSZER EC-trombomodulin-trombin kapcsolódás
trombin prokoaguláns hatása elvész PC aktíválása APC keletkezése (szerin proteáz) PS, Ca, PL jelenlétében Va, VIIIa inaktiváció Kevesebb trombin keletkezik alvadásgátlás
 PS, Ca, PL jelenlétében Va, VIIIa inaktiváció. Kevesebb trombin keletkezik. alvadásgátlás.")
20
APC REZISZTENCIA Dahlback, 1993
A leggyakoribb kongenitális trombofília Szerzett APC: aPL jelenléte esetén Kimutatás: egészséges: + APC aPTI 2x megnyúlása APC rezisztens beteg: aPTI nem nyúlik meg Háttérben legtöbbször FV mutációja FV kóros, rezisztens az APC-vel szemben

21
FV - LEIDEN 1994-ben Bertina írta le; University of Leiden
FV pontmutációja Kódoló gén az 1. kromoszómán Missence mutáció: 1961-es pozícióban G-A báziscsere G1691A 506 arg glu aminosav cserét eredményez A mutáció az APC kapcsolódási pontjánál van Mutáns FV lassabb inaktiválódás fokozott trombózis készség FVIII hasítása zavartalan FV-t is hasítja a másik 2 helyen, csak lassabb az inaktíválás enyhe trombózishajlam fokozódás Főleg más tényezők együttes jelenléte esetén Jelölés: FVR506Q, FVLeiden Autoszóm domináns öröklődés menet Ez felel az APC rezisztencia 95 %-áért

22
FV – LEIDEN GYAKORISÁG – TROMBÓZIS RIZIKÓ
Homozygota: 80x rizikó Heterozygota: 7x rizikó, általában egyéb tényező jelenléte esetén Gyakoriság: kaukázusi populációban: 6-10 % MVT-ben: 20 % fiatal, recidív trombózis: 40 % Habituális vetélés hátterében Artériás trombózis ritkább, de: fiatal, dohányos, AMI-n átesett nők: 32x rizikó, kombinált rizikó: terhesség, szülés, műtét Hemofília A: kedvező hatás, részben kompenzálja a VIII f hiányt, trombin képződés

23
Protein C deficiencia Griffin, 1981
Első magyar PC hiányos család: Domján, és mtsai: 1986 2 fenotípus 1. típus: ag és aktivitás is gyakoribb 2. típus: ag norm., aktivitás ritkább Kódoló gén a 2. kromoszómán Autoszómális domináns öröklődés Homozygota. PC < 1 % heterozygota: PC 50 % Gyakoriság: normál populáció: 0,3 %, MVT: 3,2 %

24
Klinikum Homozygota: purpura fulminans neonatorum heterozygota: 8-10 x trombózis rizikó terhesség, OAK, egyéb defektus jelentős trombózis hajlam fokozódás Kumarin okozta bőrnekrózis heparinnal átfedés kis kumarin kezdő dózis PC koncentrátum, FFP Protein C és gyulladás sepsis – purpura fulminans

25
Protein S deficiencia Comp és Esmon, 1984 PS szerepe: PC aktiválás
PS szabad formája aktív (40%) C4b-hez kötve (60%) (szerep: apoptosisban) PS defektus formái: 1. ag és funkció 2. ag norm., csak a funkció (ritkább) 3. csak a szabad frakció csökkkent Gyakoriság: PC deficienciához hasonló súlyos: ag < 1 %, purpura fulminans Mutációk többsége pontmutáció Kumarin nekrózis hátterében
 C4b-hez kötve (60%) (szerep: apoptosisban) PS defektus formái: 1. ag és funkció 2. ag norm., csak a funkció (ritkább) 3. csak a szabad frakció csökkkent. Gyakoriság: PC deficienciához hasonló súlyos: ag < 1 %, purpura fulminans. Mutációk többsége pontmutáció. Kumarin nekrózis hátterében.")
26
Prothrombin gén mutáció
Poort, 1996 2. leggyakoribb ismert trombofíliát okozó tényező Missence mutáció, G A csere FII G20210A Plazma FII szintje nő (> 115%) mechanizmus ismeretlen Homozygota: Heterozygota: 3x rizikó Prevalencia: 2 % MVT: 6-10 % heterozygota normál homozygota
 mechanizmus ismeretlen. Homozygota: Heterozygota: 3x rizikó. Prevalencia: 2 % MVT: 6-10 % heterozygota. normál. homozygota.")
27
Hyperhomocysteinemia
Leggyakoribb ok: cisztation--szintetáz hiány ritka, homozygóta súlyos trombózis hajlam Metilén-tetrahidrofolát reduktáz hiány (MTHFR) homozygóta súlyos Artériás és vénás trombózis is gyakoribb Fiatalokon atherosclerosis Lehet szerzett is (B12, B6, folsav hiány)
 homozygóta súlyos. Artériás és vénás trombózis is gyakoribb. Fiatalokon atherosclerosis. Lehet szerzett is (B12, B6, folsav hiány)")
28
Emelkedett VIII faktor szint
Egészséges populáció 10 %-ában > 150% 5x trombózis rizikó

29
A TROMBÓZIS RIZIKÓ MÉRTÉKE
Heterozygota FVLeiden: x Homozygota FVLeiden: x Heterozygota protein C deficiencia: x FII G20210A homozygota FII G20210A heterozygota x AT hiány x AT homozygota FVL (7x) + OAK (4x) x
 + OAK (4x) 28x.")
30
ÖRÖKLETES KOAGULOPÁTIÁK
VÉRZÉS TROMBÓZIS

31
Von Willebrand betegség
A leggyakoribb örökletes vérzékenység A vWf óriás glikoprotein Szerep: FVIII stabilizálás Thrombocyta adhézió Hiányában: Primer és secunder hemosztázis is zavart szenved

32
Von Willebrand betegség
Autoszómális domináns öröklődés menet vWf-t kódoló gén a 12. kromoszómán MK, EC, placenta sejtjeiben expresszálódik Mutáció következtében nem termelődik fehérje, vagy kóros fukció A 22. kromoszómán kisebb méretű pseudogén

33
Gyakoriság Kezelést igénylő betegség: 1/10.000
A hordozók többsége tünetmentes 1. típus (70%): mennyiségi csökkenés általában heterozygota 2. típus: funkció zavar, heterogén csoport, heterozygoták 3. típus: vWf hiány, súlyos vérzékenység 2-3. típusnak autoszomális recesszív formája is van
: mennyiségi csökkenés általában heterozygota. 2. típus: funkció zavar, heterogén csoport, heterozygoták. 3. típus: vWf hiány, súlyos vérzékenység típusnak autoszomális recesszív formája is van.")
34
HEMOFÍLIÁK Hemofília A FVIII hiány hemofília B FIX hiány, diszfukció
FVIII és F IX génje az X kromoszómán Nők hordozók (heterozygoták) Férfiak betegednek meg (hemizygoták) Ritkán a nők is lehetnek betegek (gén inaktiváció, mozaicizmus, hordozó anya-beteg apa gyereke) Klinikailag nem különíthető el
 Férfiak betegednek meg (hemizygoták) Ritkán a nők is lehetnek betegek (gén inaktiváció, mozaicizmus, hordozó anya-beteg apa gyereke) Klinikailag nem különíthető el.")
35
A BETEGSÉG SÚLYOSSÁGA A faktor szinttől függenek Súlyos: < 1%
Kp: % Enyhe: > 5 %

36
Szerep: FX aktiválása

37
ÖRÖKLŐDÉS

38
„KIRÁLYI BETEGSÉG” Viktória királynő Alekszisz (II. Miklós cár fia)
")
40
HEMOFÍLIA A Gyakoriság: 1:10.000 5x gyakoribb, mint „B”
30 %-ban új mutáció Genetikai háttér: Pontmutáció, inverzió, deléció Heterogén eltérések Súlyos esetek 50 %-a: intron 22 inverzió Southern blott-tal kimutatható Többi eltérés indirekt molekuláris genetikai vizsgálatokkal

41
HEMOFÍLIA B Kimutatás: indirekt genetikai vizsgálatok FIX génje rövidebb, szekvencia analízis

42
Hordózó állapot, prenatális diagnosztika lehetséges

43
KLINIKUM Izületi bevérzések

44
Nagy, lapszerint vérzések
Epiduralis hematoma „re bleeding”

45
Diagnózis aPTI megnyúlt
Normál plazmával korrigálható (ha nem: gátlótest)
")
46
Differenciál diagnózis
vWD FXI hiány Szerzett gátlótestes hemofília „B” K vitamin hiány Kumarin kezelés Májbetegség APS FIX elleni antitest

47
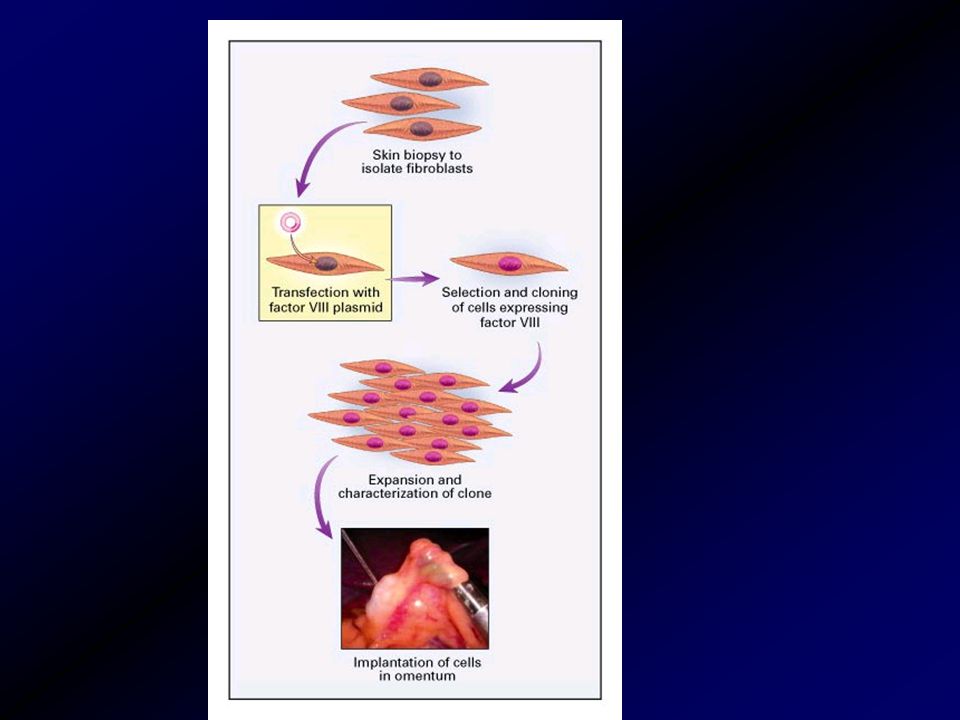
A jövő terápiája gén terápia
Az alvadási faktor génjét májsejtekbe viszik be

49
A THROMBOCYTOPATHIÁK VELESZÜLETETT BETEGSÉGEI
Normális thrombocytaszám Megnyúlt vérzési idő Klinikailag bőr, nyálkahártya vérzések Ritka betegségek Általában autoszomális recesszív öröklődésű géndefektusok Dg: thrombocyta funkciós labor vizsgálatok Vérlemezkék morfológiai eltérései

50
VIZSGÁLATOK Vérzési idő Kóros morfológia (óriás, kicsi thrombocyták)
Thrombocyta aggregáció Elektromikroszkópos vizsgálat Thrombocyta membrán receptor vizsgálata PFA-100 – platelet function analyzer készülék

51
AZ EGYES KÓRKÉPEKBEN Az Adhézió Aggregáció Szekréció
Intracelluláris szignál átvitel Prokoaguláns aktivitás zavara

52
Az adhézió zavara Bernard-Soulier szindróma Óriás thrombocyták
GPIb-IX-V komplex képzési zavara A kóros T nem tud kitapadni a subendoteliális kollagénhez Pseudo-von-Willebrand –betegség GPIb receptor fokozottan köti a vWf-t, a keringésben csökken a vWf szint Kollagén receptor hiány GPVI és GPIa-IIa hiánya

53
Az aggregáció zavara Glanzmann thrombasthenia autoszómális recesszív öröklésmenet aggregáció hiánya súlyos vérzések GPIIb-IIIa receptorokat kódoló gének mutációi

54
A szekréció zavarai Enyhe, vagy mérsékelt súlyosságú vérzések
Thrombocyta granulumok hiánya, szekréciós mechanizmus rendellenességei Gray platelet szindróma granulum hiánya – 40 eset Quebec thrombocyta betegség Storage-pool disease Hermansky-Pudlak szindróma Chediak-Higashi szindróma fvs kemotaktikus és baktericid aktivitás károsodott, infekciók, albinizmus Wiskott-Aldrich szindróma X-hez kötött, ekzema, immundeficiencia, kis thrombocyták

55
Bernard Soulier Wiskott-Aldrich Gray platelet

56
KÖSZÖNÖM MEGTISZTELŐ FIGYELMÜKET






















 kialakulása Genetikai, Sejt- és Immunbiológiai Intézet Falus András.>")
