Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
a tumor-specifikus immunitás kiváltása
Tumor immunoterápia: a tumor-specifikus immunitás kiváltása

2
TUMOR Tumor (neoplázia): növekedési kontrol (immunosurveillance) alól elszabadult sejtek csoportja Benignus (jóindulatú) korlátozott növekedés Malignus (rosszindulatú) korlátlan növekedés (rák) Metastasis: áttét Carcinoma: embrionális ektoderma, vagy endoderma eredetű Sarcoma: mezodermális kötőszövet (szövet, csont) „ Leukémia: hematopoietikus sejt eredetű Limfóma: csontvelői hematopoetikus sejtekből A sejtek malignus transzformációja: Kémiai karcinogén anyagok Besugárzás mutáció , transzformáció Vírusok
 korlátozott növekedés. Malignus (rosszindulatú) korlátlan növekedés (rák) Metastasis: áttét. Carcinoma: embrionális ektoderma, vagy endoderma eredetű. Sarcoma: mezodermális kötőszövet (szövet, csont) „ Leukémia: hematopoietikus sejt eredetű. Limfóma: csontvelői hematopoetikus sejtekből. A sejtek malignus transzformációja: Kémiai karcinogén anyagok. Besugárzás mutáció , transzformáció. Vírusok.")
3
A veleszületett és az adaptív immunválasz elemei.
Nature Reviews Cancer 4; (2004); CYTOKINES IN CANCER PATHOGENESIS AND CANCER THERAPY Figure 1 | The innate and adaptive immune response. The innate immune response functions as the first line of defence against infection. It consists of soluble factors, such as complement proteins, and diverse cellular components including granulocytes (basophils, eosinophils and neutrophils), mast cells, macrophages, dendritic cells and natural killer cells. The adaptive immune response is slower to develop, but manifests as increased antigenic specificity and memory. It consists of antibodies, B cells, and CD4+ and CD8+ T lymphocytes. Natural killer T cells and T cells are cytotoxic lymphocytes that straddle the interface of innate and adaptive immunity. A veleszületett és az adaptív immunválasz elemei.
; CYTOKINES IN CANCER PATHOGENESIS AND CANCER THERAPY. Figure 1 | The innate and adaptive immune response. The innate immune response functions as the first line of defence against infection. It consists of soluble factors, such as complement proteins, and diverse cellular components including granulocytes (basophils, eosinophils and neutrophils), mast cells, macrophages, dendritic cells and natural killer cells. The adaptive immune response is slower to develop, but manifests as increased antigenic specificity and memory. It consists of antibodies, B cells, and CD4+ and CD8+ T lymphocytes. Natural killer T cells and T cells are cytotoxic lymphocytes that straddle the interface of innate and adaptive immunity. A veleszületett és az adaptív immunválasz elemei.")
4
A tumor felismerésének közvetett és közvetlen útjai
Figure 2 | Direct and indirect pathways of cancer recognition. a | Innate immune cells recognize cancers through germ-line-encoded pattern-recognition receptors and other cell-surface molecules. Cancers express various stress-induced genes such as MICA and MICB that trigger NKG2D receptors on natural killer (NK) cells, macrophages and some cytotoxic T lymphocytes (CTLs). NK cells also use a combination of inhibitory and activating receptors such as the killer-cell immunoglobulin-like receptors (KIRs) to detect the loss of major histocompatibility complex (MHC) class I expression on cancers. Dendritic cells (DCs) phagocytose apoptotic tumours through CD36 and v 5. Macrophages and DCs also use scavenger receptors and CD91 to ingest heat-shock proteins (HSPs) complexed with tumour-derived peptides that are released from necrotic cancer cells. b | Adaptive immune cells are initially stimulated to recognize cancer cells through cross-priming by DCs. As tumour cells do not express co-stimulatory molecules that are important for T-cell activation, they generally cannot prime cellular responses efficiently. Instead, DCs capture dying tumour cells or debris, migrate to regional lymph nodes and process the tumour-derived material onto CD1D, for presentation to NKT cells (not shown), and MHC class I and class II molecules for presentation to CD4+ and CD8+ T cells. Mature DCs express co-stimulatory molecules such as B7-1 and B7-2 to enhance T-cell activation. DCs secrete IL-12 and IL-18 to promote T helper 1 (TH1) CD4+ T-cell responses and cytotoxic CD8+ T cells. Activated CD4+ T cells and NKT cells express CD40 ligand, which further stimulates DC maturation through CD40 signalling. CD4+ T cells and DCs also help trigger B cells to produce antibodies that are reactive with tumour proteins.
 cells, macrophages and some cytotoxic T lymphocytes (CTLs). NK cells also use a combination of inhibitory and activating receptors such as the killer-cell immunoglobulin-like receptors (KIRs) to detect the loss of major histocompatibility complex (MHC) class I expression on cancers. Dendritic cells (DCs) phagocytose apoptotic tumours through CD36 and v 5. Macrophages and DCs also use scavenger receptors and CD91 to ingest heat-shock proteins (HSPs) complexed with tumour-derived peptides that are released from necrotic cancer cells. b | Adaptive immune cells are initially stimulated to recognize cancer cells through cross-priming by DCs. As tumour cells do not express co-stimulatory molecules that are important for T-cell activation, they generally cannot prime cellular responses efficiently. Instead, DCs capture dying tumour cells or debris, migrate to regional lymph nodes and process the tumour-derived material onto CD1D, for presentation to NKT cells (not shown), and MHC class I and class II molecules for presentation to CD4+ and CD8+ T cells. Mature DCs express co-stimulatory molecules such as B7-1 and B7-2 to enhance T-cell activation. DCs secrete IL-12 and IL-18 to promote T helper 1 (TH1) CD4+ T-cell responses and cytotoxic CD8+ T cells. Activated CD4+ T cells and NKT cells express CD40 ligand, which further stimulates DC maturation through CD40 signalling. CD4+ T cells and DCs also help trigger B cells to produce antibodies that are reactive with tumour proteins.")
5
Tumorsejtek immun-átszerkesztése és szelekciója
Cancer immunoediting encompasses three process. (a) Elimination corresponds to immunosurveillance. (b) Equilibrium represents the process by which the immune system iteratively selects and/or promotes the generation of tumor cell variants with increasing capacities to survive immune attack. (c) Escape is the process wherein the immunologically sculpted tumor expands in an uncontrolled manner in the immunocompetent host. In a and b, developing tumor cells (blue), tumor cell variants (red) and underlying stroma and nontransformed cells (gray) are shown; in c, additional tumor variants (orange) that have formed as a result of the equilibrium process are shown. Different lymphocyte populations are as marked. The small orange circles represent cytokines and the white flashes represent cytotoxic activity of lymphocytes against tumor cells. fejlődő tumor sejtek túlélő tumor sejt variánsok Nem transzformált sejtek Ellenőrzés alól elszabadult osztódó tumorsejtek, újabb variánsok
 Elimination corresponds to immunosurveillance. (b) Equilibrium represents the process by which the immune system iteratively selects and/or promotes the generation of tumor cell variants with increasing capacities to survive immune attack. (c) Escape is the process wherein the immunologically sculpted tumor expands in an uncontrolled manner in the immunocompetent host. In a and b, developing tumor cells (blue), tumor cell variants (red) and underlying stroma and nontransformed cells (gray) are shown; in c, additional tumor variants (orange) that have formed as a result of the equilibrium process are shown. Different lymphocyte populations are as marked. The small orange circles represent cytokines and the white flashes represent cytotoxic activity of lymphocytes against tumor cells. fejlődő tumor sejtek. túlélő tumor sejt variánsok. Nem transzformált sejtek. Ellenőrzés alól elszabadult osztódó tumorsejtek, újabb variánsok.")
6
NKT sejtek NK1.1+, abTCR+ sejtek, CD1d-függő, MHC-tól független
korlátozott repertoár: invariáns alfa lánc, béta lánc:Vb8.2,Vb2,Vb7; Vb11 (human) Glycolipidek, lipidek (saját és mikrobiális) hidrofil fejének felismerése (a-glycosphingolipid) a – galactosyl ceramide: IL-4, IL-10, IL-13, IFNg –t termel –> regulator funkció Tumor sejtek pusztítása in vitro! Figure 1. Structure of the synthetic α-galactosylceramide.Sphingosine long chain base is shown in green, acyl chain in blue and galactose in red. Galactose is in alpha configuration (see also figure 3). Carbons 1 to 4 on sphingosine are indicated with numbers. In the acyl chain, the alpha carbon position immediately following the carboxyl group is indicated.
 Glycolipidek, lipidek (saját és mikrobiális) hidrofil fejének felismerése (a-glycosphingolipid) a – galactosyl ceramide: IL-4, IL-10, IL-13, IFNg –t termel –> regulator funkció. Tumor sejtek pusztítása in vitro! Figure 1. Structure of the synthetic α-galactosylceramide.Sphingosine long chain base is shown in green, acyl chain in blue and galactose in red. Galactose is in alpha configuration (see also figure 3). Carbons 1 to 4 on sphingosine are indicated with numbers. In the acyl chain, the alpha carbon position immediately following the carboxyl group is indicated.")
7
A tumor immunsejtek által történő elpusztításának modelje
. A proposed model for the elimination phase of the cancer immunoediting process. (a) The initiation of the response in which lymphocytes that participate in innate immunity (NKT, NK and T cells) recognize transformed cells that have accumulated above a threshold that has yet to be defined and are stimulated to produce IFN- . (b) The initial IFN- starts a cascade of innate immune reactions that involve (i) the induction of chemokines, including the angiostatic chemokines (CXCL10 (1P10), CXCL9 (MIG) AND CXCL11 (I-TAC)) that block neovascularization in the tumor and that also effect the recruitment of NK cells, dendritic cells, macrophages and other immune effector cells to the tumor site; (ii) an antiproliferative action of IFN- on the developing tumor and (iii) the activation of cytocidal activity in macrophages and NK cells entering the tumor. These events result in some tumor cell death by both immunologic and nonimmunologic mechanisms. Dead tumor cells or tumor cell debris (blue squares) are ingested by dendritic cells and are trafficked to the draining lymph node. (c) Tumor growth is kept in check by the cytocidal activities of NK cells and activated macrophages while CD4+ and CD8+ T cells that are specific for tumor antigens develop in the draining lymph node. (d) Tumor-specific CD4+ and CD8+ T cells home to the tumor along a chemokine gradient where they recognize and destroy tumor cells expressing distinctive tumor antigens. Tumor cells (blue); nontransformed cells (gray); dead tumor cells (white to gray gradient circles surrounded by a dashed black line); lymphocytes, dendritic cells (DC) and macrophages (Mac) are marked and colored appropriately.
 The initiation of the response in which lymphocytes that participate in innate immunity (NKT, NK and T cells) recognize transformed cells that have accumulated above a threshold that has yet to be defined and are stimulated to produce IFN- . (b) The initial IFN- starts a cascade of innate immune reactions that involve (i) the induction of chemokines, including the angiostatic chemokines (CXCL10 (1P10), CXCL9 (MIG) AND CXCL11 (I-TAC)) that block neovascularization in the tumor and that also effect the recruitment of NK cells, dendritic cells, macrophages and other immune effector cells to the tumor site; (ii) an antiproliferative action of IFN- on the developing tumor and (iii) the activation of cytocidal activity in macrophages and NK cells entering the tumor. These events result in some tumor cell death by both immunologic and nonimmunologic mechanisms. Dead tumor cells or tumor cell debris (blue squares) are ingested by dendritic cells and are trafficked to the draining lymph node. (c) Tumor growth is kept in check by the cytocidal activities of NK cells and activated macrophages while CD4+ and CD8+ T cells that are specific for tumor antigens develop in the draining lymph node. (d) Tumor-specific CD4+ and CD8+ T cells home to the tumor along a chemokine gradient where they recognize and destroy tumor cells expressing distinctive tumor antigens. Tumor cells (blue); nontransformed cells (gray); dead tumor cells (white to gray gradient circles surrounded by a dashed black line); lymphocytes, dendritic cells (DC) and macrophages (Mac) are marked and colored appropriately.")
8
NK sejtek és a tumorra adott immunválasz
Nature Reviews Cancer 2; (2002); doi: /nrc928 NEW ASPECTS OF NATURAL-KILLER-CELL SURVEILLANCE AND THERAPY OF CANCER < previous next > NK sejtek és a tumorra adott immunválasz NK cells and immune responses to tumour cells. The diagram shows a hypothetical scheme of the potential role of natural-killer (NK) cells in tumour immune surveillance and in the network of immune cells that respond to tumours. NK cells might intially recognize certain 'stress' or 'danger' signals that are produced by tumours. Both NK cells and cytotoxic T cells (CTLs) are important mediators of antitumour immunity, as they are ultimately responsible for the destruction of the malignant cells. NK cells can influence the development of adaptive T- and B-cell immune responses that constitute specific immunity and immunological memory to tumours and pathogens. NK-cell lysis of cancer cells could provide tumour antigens for dendritic cells (DCs), which induce them to mature and present antigen (Ag) to CTLs in lymph nodes. Cytokines, such as interferon (IFN)- , which are produced by activated NK cells, activate CTL and helper T-cell (CD4+) responses. This leads to the proliferation of helper T cells and cytokine production. Activated NK1.1+ T (NKT) cells can also induce the antitumour activity of NK cells. Cytokines that are produced by NK cells might also regulate B-cell production of antitumour antibodies (Abs).
; doi: /nrc928 NEW ASPECTS OF NATURAL-KILLER-CELL SURVEILLANCE AND THERAPY OF CANCER. < previous next > NK sejtek és a tumorra adott immunválasz NK cells and immune responses to tumour cells. The diagram shows a hypothetical scheme of the potential role of natural-killer (NK) cells in tumour immune surveillance and in the network of immune cells that respond to tumours. NK cells might intially recognize certain stress or danger signals that are produced by tumours. Both NK cells and cytotoxic T cells (CTLs) are important mediators of antitumour immunity, as they are ultimately responsible for the destruction of the malignant cells. NK cells can influence the development of adaptive T- and B-cell immune responses that constitute specific immunity and immunological memory to tumours and pathogens. NK-cell lysis of cancer cells could provide tumour antigens for dendritic cells (DCs), which induce them to mature and present antigen (Ag) to CTLs in lymph nodes. Cytokines, such as interferon (IFN)- , which are produced by activated NK cells, activate CTL and helper T-cell (CD4+) responses. This leads to the proliferation of helper T cells and cytokine production. Activated NK1.1+ T (NKT) cells can also induce the antitumour activity of NK cells. Cytokines that are produced by NK cells might also regulate B-cell production of antitumour antibodies (Abs).")
9
NK sejtek effektor funkciói, amelyek elpusztítják a tumorsejteket.
NK-cell effector functions that eliminate tumour cells. Schematic illustrations of effector functions of natural-killer (NK) cells that control tumour cells. NK cells can use the perforin/ granzyme-containing granule exocytosis pathway (a), the death-receptor-ligand pathway (b) or the nitric oxide pathway (c) to kill tumour cells. NK cells can also produce cytokines such as interferon (IFN)- that restrict tumour angiogenesis and stimulate adaptive immunity (not shown). a | Within a few minutes of effector–tumour-cell interaction (conjugation), cytotoxic granules become re-oriented towards the tumour cell, and their contents are secreted into the intercellular cleft between effector and tumour cells in a calcium-dependent manner. These granules contain perforin, which disturbs the tumour-cell membrane and allows the entry of serine proteases known as granzymes. The two most-abundant granzymes, A and B, have been implicated in mediating apoptosis of target cells through caspase-dependent and -independent pathways. Signalling through activating receptors induce this activity, whereas inhibitory receptors prevent it. b | Some NK cells express FAS ligand (FASL) or tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL). These bind to and activate their receptors, FAS and TRAIL receptor (TRAILR), respectively, which are expressed by tumour cells. This interaction induces tumour-cell apoptosis. The protein c-FLIP (FLICE inhibitory protein) blocks apoptosis induction through this pathway. c | NK cells secrete various effector molecules, such as IFN- . IFN- exerts antitumour functions in many different ways. Nitric oxide (NO) is one of the most powerful effector molecules in the immune system, and can regulate the cytotoxic function of NK cells against tumour cells. It remains unclear in this situation whether NK cells or another cell type produces NO. MHC, major histocompatibility complex.
 cells that control tumour cells. NK cells can use the perforin/ granzyme-containing granule exocytosis pathway (a), the death-receptor-ligand pathway (b) or the nitric oxide pathway (c) to kill tumour cells. NK cells can also produce cytokines such as interferon (IFN)- that restrict tumour angiogenesis and stimulate adaptive immunity (not shown). a | Within a few minutes of effector–tumour-cell interaction (conjugation), cytotoxic granules become re-oriented towards the tumour cell, and their contents are secreted into the intercellular cleft between effector and tumour cells in a calcium-dependent manner. These granules contain perforin, which disturbs the tumour-cell membrane and allows the entry of serine proteases known as granzymes. The two most-abundant granzymes, A and B, have been implicated in mediating apoptosis of target cells through caspase-dependent and -independent pathways. Signalling through activating receptors induce this activity, whereas inhibitory receptors prevent it. b | Some NK cells express FAS ligand (FASL) or tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL). These bind to and activate their receptors, FAS and TRAIL receptor (TRAILR), respectively, which are expressed by tumour cells. This interaction induces tumour-cell apoptosis. The protein c-FLIP (FLICE inhibitory protein) blocks apoptosis induction through this pathway. c | NK cells secrete various effector molecules, such as IFN- . IFN- exerts antitumour functions in many different ways. Nitric oxide (NO) is one of the most powerful effector molecules in the immune system, and can regulate the cytotoxic function of NK cells against tumour cells. It remains unclear in this situation whether NK cells or another cell type produces NO. MHC, major histocompatibility complex.")
10
Replikációs életszakasz Gén expresszió változás
Az önfenntartó sejtszaporodáshoz vezető molekuláris útvonalak Sejtciklus Replikációs életszakasz Gén expresszió változás Apoptózis Cancer arises from the stepwise accumulation of genetic changes that confer upon an incipient neoplastic cell the properties of unlimited, self-sufficient growth and resistance to normal homeostatic regulatory mechanisms. Advances in human genetics and molecular and cellular biology have identified a collection of cell phenotypes — the main destinations in the subway map below — that are required for malignant transformation1. Specific molecular pathways (subway lines) are responsible for programming these behaviours. Although the connections between cancer-cell wiring and function remain incompletely explored and specified — hence the many lines under construction — the broad outlines of the molecular circuitry of the cancer cell can now be sketched. Further advances in understanding these pathways and their interconnections will accelerate the development of molecularly targeted therapies that promise to change the practice of oncology. Anyagcsere
 are responsible for programming these behaviours. Although the connections between cancer-cell wiring and function remain incompletely explored and specified — hence the many lines under construction — the broad outlines of the molecular circuitry of the cancer cell can now be sketched. Further advances in understanding these pathways and their interconnections will accelerate the development of molecularly targeted therapies that promise to change the practice of oncology. Anyagcsere.")
11
Tumor kialakulásában szerepet játszó gének:
tumor gének 291 tumor gént azonosítottak, ez több mint 1 %-a a teljes genomnak Ezek 90 %-a szomatikus mutáción esett át, 20 % embrionális mutációt, 10 % mindkettőt tartalmaz Kromoszóma transzlokáció a leggyakoribb – kiméra gén A legtöbb tumor gént azonosították leukémia, limfóma és szarkóma esetében. Ezek csak 10 %-át adják az összes tumoros betegségeknek A legközönségesebb domén, amely a tumor gének által kódolt: protein kináz 2004-es felmérés

12
A tumorspecifikus antigén kialakulása
karcinogén „privát antigének”

13
Kromoszóma transzlokáció Burkitt limfómában
EBV fertőzés: c-myc az áthelyeződik az Ig H lánc enhancer régiójához – B sejtek korlátlan növekedése

14
a sejttranszformációt kiváltó fehérjét kódoló gén
Onkogén: a sejttranszformációt kiváltó fehérjét kódoló gén 1910: Rous sarcoma virus: v-src (c-src) 1966 Nobel díj (csirke szarkóma sejtek szűrlete RNS vírust tartalmaz) Normális sejtekben: proto-onkogének, a sejtproliferációt, sejtciklust, túlélést, illetve apoptózist szabályozó fehérjék Mutáció, transzlokáció következtében megváltozik: aktivitásuk, mennyiségük, funkciójuk
 1966 Nobel díj. (csirke szarkóma sejtek szűrlete RNS vírust tartalmaz) Normális sejtekben: proto-onkogének, a sejtproliferációt, sejtciklust, túlélést, illetve apoptózist szabályozó fehérjék. Mutáció, transzlokáció következtében megváltozik: aktivitásuk, mennyiségük, funkciójuk.")
15
Immortalizáció: hallhatatlan sejtek
Initiation (mutáció) – transzformáció- Promotion: osztódási képesség fokozódik Kiváltó okok: - Xeroderma pigmentosum: UV specifikus endonuclease (DNA repair) hiánya bőrrák - Vírusok: (15 %), DNS vírusok - RNS vírusok (citoplazmában) SV40 (papilloma virus), polyoma - retrovírusok · Oncogen: rák-gén -> a transzformációt kiváltó fehérjét kódoló gén Rous sarcoma virus: v-src (c-src) · Proto-onkogens (cellular oncogens): sejtosztódást ellenörzö fehérjék csoportja
 – transzformáció- Promotion: osztódási képesség fokozódik. Kiváltó okok: - Xeroderma pigmentosum: UV specifikus endonuclease (DNA repair) hiánya bőrrák. - Vírusok: (15 %), DNS vírusok - RNS vírusok (citoplazmában) SV40 (papilloma virus), polyoma - retrovírusok. · Oncogen: rák-gén -> a transzformációt kiváltó fehérjét kódoló gén. Rous sarcoma virus: v-src (c-src) · Proto-onkogens (cellular oncogens): sejtosztódást ellenörzö fehérjék csoportja.")
16
Onkogének csoportosítása, funkciója:
sejtosztódást indukáló (GF or GFR, szignál fehérje, transzkripciós faktor) sejtosztódást gátló : tumor szuppresszor gének (anti-onkogének) : Rb, p53 mutációja programmozott sejthalál (apoptózis) szabályozó fehérjéi: bcl2 Protoonkogén carcinogén v. vírus hatására Onkogén
 sejtosztódást gátló : tumor szuppresszor gének (anti-onkogének) : Rb, p53 mutációja. programmozott sejthalál (apoptózis) szabályozó fehérjéi: bcl2. Protoonkogén. carcinogén v. vírus hatására. Onkogén.")
17
Sejt ciklus: tumor szuppresszor fehérjék ellenőrzése alatt áll
G1 fázisban külső+belső jelek alapján döntenek a nyugvó állapot vagy a sejtciklusba lépés között További ellenőrzési pontok későbbi fázisban Tumor szuppressziós útvonalak gátoltak a tumorsejtekben retonoblastoma (RB) út: E2F transzkripciós faktorok által ellenőrzött gének (sejtciklus) expresszióját gátolja, foszforilációtól függően inaktiválódik, a cyclin dependens kinázok szabályozzák P53 út: apoptózist aktivál, gátolja a sejtciklusba lépést (CDK inhibitort aktivál) Normális sejtben a RAS útvonal tartós aktiválása aktiválja az RB és p53 utat-> befagyasztja a sejtciklust - védekező mechanizmus TGFb út: ser/thr kinázok aktiválása -> SMAD transzkripciós faktorok aktiválása sejtciklust szabályozó gének (CDK gátlók, Myc onkogén).
 út: E2F transzkripciós faktorok által ellenőrzött gének (sejtciklus) expresszióját gátolja, foszforilációtól függően inaktiválódik, a cyclin dependens kinázok szabályozzák. P53 út: apoptózist aktivál, gátolja a sejtciklusba lépést (CDK inhibitort aktivál) Normális sejtben a RAS útvonal tartós aktiválása aktiválja az RB és p53 utat-> befagyasztja a sejtciklust - védekező mechanizmus. TGFb út: ser/thr kinázok aktiválása -> SMAD transzkripciós faktorok aktiválása sejtciklust szabályozó gének (CDK gátlók, Myc onkogén).")
18
A tumor és az immunrendszer

19
Tumorsejtek támadása a gazdaszervezet T sejtjei ellen
Fig. 1. The Fas counterattack. Cancer cells are frequently resistant to apop- tosis mediated through Fas. This might be a result of downregulation of Fas or the release of soluble Fas, or abnormalities in the level of several proteins involved in the signal transduction cascade. Downregulation of caspase 1, Bax or Bak, and upregulation of FLIP, FAP-1 or Bcl2, have all been impli- cated as potential mechanisms of Fas resistance in various cancer cells. In addition, mutations have been identified in some components of the path- way, including Fas itself and caspase 8. There is evidence that some onco- gene and tumor suppressor gene mutations, commonly found in tumors, could potentially impair Fas signaling (p53 and Ras), or cooperate with Fas resistance (c-Myc) in certain tumor cells. Many cancer cells also ex- press FasL, and can therefore counterattack and kill Fas-sensitive tumor- infiltrating lymphocytes (TILs). Systemic consequences of the counter- attack are speculative, but might include tolerance to tumor antigens. Abbreviations: Bcl2, B cell lymphoma leukemia 2 protein; Bax, Bcl2- antagonist x protein; Bak, Bcl2-associated killer protein; caspase, cysteinyl aspartate-specific protease; FLIP, FLICE (caspase 8) inhibitory protein; FAP-1, Fas-associated phosphatase 1; FasL, Fas ligand.
, or cooperate with. Fas resistance (c-Myc) in certain tumor cells. Many cancer cells also ex- press FasL, and can therefore counterattack and kill Fas-sensitive tumor- infiltrating lymphocytes (TILs). Systemic consequences of the counter- attack are speculative, but might include tolerance to tumor antigens. Abbreviations: Bcl2, B cell lymphoma leukemia 2 protein; Bax, Bcl2- antagonist x protein; Bak, Bcl2-associated killer protein; caspase, cysteinyl. aspartate-specific protease; FLIP, FLICE (caspase 8) inhibitory protein; FAP-1, Fas-associated phosphatase 1; FasL, Fas ligand.")
20
Tumorsejtek megszöknek a T sejt felimerés elől
Tumor sejtekben az antigén processzálása gátolt: MHCI, TAP, proteaszoma FasL expresszió növelése T sejtek apoptózisa, Gátló citokinek termelése (IL-10, TGFβ) Mechanisms of tumour-cell escape from T-cell recognition. Recent progress in our understanding of the interactions of tumour cells with the host’s immune system has led to the realization that tumour cells as well as other cells in the tumour’s microenvironment devise multiple ways to evade being recognized and lysed by activated T cells. There are at least two ways by which the tumour cell directly avoids T-cell recognition. Tumour cells can downregulate or genetically alter components of their antigen processing and presentation machinery including major histocompatibility (MHC) class I molecules, the TAP (transporter associated with antigen processing) transporter subunits, and subunits of the proteasome. The latter cytoplasmic enzyme complex normally digests whole cellular proteins into the peptide fragments that bind to MHC class I antigens. Tumour cells can also upregulate FAS ligand (FasL) and induce T-cell apoptosis via FAS on T cells. Endothelial cells and other cells within the microenvironment can also contribute to immune cell inhibition via the secretionof immune inhibitory cytokines.
 Mechanisms of tumour-cell escape from T-cell recognition. Recent progress in our understanding. of the interactions of tumour cells with the host’s immune system has led to the realization that tumour cells as well as other cells in the tumour’s microenvironment devise multiple ways to evade being recognized and lysed by activated T cells. There are at least two ways by which the tumour cell directly avoids T-cell recognition. Tumour cells can downregulate or genetically alter components of their antigen processing and presentation machinery including major histocompatibility (MHC) class I molecules, the TAP (transporter associated with antigen processing) transporter subunits, and subunits of the proteasome. The latter cytoplasmic enzyme complex normally digests whole cellular proteins into the peptide fragments that bind to MHC class I antigens. Tumour cells can also upregulate FAS ligand (FasL) and induce T-cell apoptosis via FAS on T cells. Endothelial cells and other cells within the microenvironment can also contribute to immune cell inhibition via the secretionof immune inhibitory cytokines.")
21
Tumor „megszökése” az immunrendszer elől:
· alacsony MHCI expresszió, csökkent prezentáció · nincs MHCII expresszió - TH sejteket nem aktivál · kostimulátor molekulák (B7) hiánya · tumorsejt terméke elnyomja immunválaszt (TGF, IL-10) · a gazdaszervezet toleranciája · mutáns tumor sejtek elszaporodása, amelyek nem expresszálnak peptid- MHCI komplexet · antigén moduláció: tumor antigének elveszitése, vagy elrejtése ellenanyagok által · rezisztens tumor kialakulása · tumor antigének elfedése sziálsav tartalmu mukopoliszaharidok által - „antigen masking”
 hiánya. · tumorsejt terméke elnyomja immunválaszt (TGF, IL-10) · a gazdaszervezet toleranciája. · mutáns tumor sejtek elszaporodása, amelyek nem expresszálnak peptid- MHCI komplexet. · antigén moduláció: tumor antigének elveszitése, vagy elrejtése ellenanyagok által. · rezisztens tumor kialakulása. · tumor antigének elfedése sziálsav tartalmu mukopoliszaharidok által - „antigen masking")
22
A tumor antigéneket felismerheti mind a természetes, mind a szerzett immunrendszer.
A gazdaszervezet citokin termelése elnyomhatja a tumor képződést az infekció, gyulladás és az immunválasz szabályozása révén A tumor sejtek felhasználhatják a gazda citokinjeit a szaporodás elősegítésére, az apoptózistól való megmenekülésre és a szétterjedésre Citokinek szisztémás alkalmazása anti-tumor hatású, de súlyos toxikus mellékhatásokkal jár, mint pl. infekció, ez behatárolja az alkalmazást. Tumor sejteket genetikai manipulációval módosítani lehet immunstimuláló citokinek termelésére, ezek tumor vakcinák alapjait képezhetik CTLA-4 blokkoló receptor gátlása ellenanyaggal ígéretes stratégia a tumor vakcina hatékonyságának növelésére, de rizikó faktor –saját antigénekkel szembeni toleranciát is elnyomhatja.

23
CÉL: a gazdaszervezet tumorspecifikus immunválaszának fokozása
IMMUNTERÁPIA CÉL: a gazdaszervezet tumorspecifikus immunválaszának fokozása Tumor antigének: Tumor sejtekben a mutációk gyakorisága nagy => fehérje expresszió a normálistól eltér Oncogén virusok által indukált tumor: vírus fehérjék expressziója (CTL aktiválás) Abnormális szerkezetű lipidek, szénhidrátok szintézise (B sejtek aktiválása) Tumor specifikus antigének -normális sejten nincs Tumor asszociált antigének - normális sejten is van -tolerogén
 Abnormális szerkezetű lipidek, szénhidrátok szintézise (B sejtek aktiválása) Tumor specifikus antigének -normális sejten nincs. Tumor asszociált antigének - normális sejten is van -tolerogén.")
25
A tumor és az immunrendszer egymásrahatása - a tumor ellenes immunválasz kialakulása
CD8+ Tc-sejt Tumor-specifikus CTL indukció Citokinek Tumor antigének szeparálása Dendritikus sejtek feltöltése Tumor-specifikus CTL Genetikai módosítás (MHCII expresszió)
")
26
A tumorsejtek transzfektálása B7 –tel, vagy GM-CSF –el növeli a tumor immunogenitását
Transfection of tumors with the gene for B7 or for GM-CSF enhances tumor immunogenicity. A tumor that does not express co-stimulatory molecules will not induce an immune response, even though it might express tumor rejection antigens (TRAs), because naive CD8 T cells specific for the TRA cannot be activated by the tumor. The tumor therefore grows progressively in normal mice and eventually kills the host (top panels). If such tumor cells are transfected with a co-stimulatory molecule, such as B7, TRA-specific CD8 T cells now receive both signal 1 and signal 2 from the same cell (see Section 8-5) and can therefore be activated (center panels). The same effect can be obtained by transfecting the tumor with the gene encoding GM-CSF, which attracts and stimulates the differentiation of dendritic cell precursors (bottom panels). Both these strategies have been tested in mice and shown to elicit memory T cells, although results with GM-CSF are more impressive. Because TRA-specific CD8 cells have now been activated, even the original B7-negative or GM-CSF negative tumor cells can be rejected.
, because naive CD8 T cells specific for the TRA cannot be activated by the tumor. The tumor therefore grows progressively in normal mice and eventually kills the host (top panels). If such tumor cells are transfected with a co-stimulatory molecule, such as B7, TRA-specific CD8 T cells now receive both signal 1 and signal 2 from the same cell (see Section 8-5) and can therefore be activated (center panels). The same effect can be obtained by transfecting the tumor with the gene encoding GM-CSF, which attracts and stimulates the differentiation of dendritic cell precursors (bottom panels). Both these strategies have been tested in mice and shown to elicit memory T cells, although results with GM-CSF are more impressive. Because TRA-specific CD8 cells have now been activated, even the original B7-negative or GM-CSF negative tumor cells can be rejected.")
27
TUMOR ANTIGÉNEK Tumor ellen gazdaszervezet immunválasszal reagál: limfocita függő Specifikus tumor ellenes ellenanyagok memória van „Immunsurveilance” - tumor elpusztítására tumor specifikus antigének (TSA) - csak az adott tumorra jellemző MHC-hez kötődött peptidek tumorhoz asszociált antigének (TAA) - embrionális gének termékei- mennyiségi növekedés 50x- 100x Tumor antigének izolálása: cDNS könyvtárból - CTL segítségével, tumor sejtek felszínéről savas elucióval
 - csak az adott tumorra jellemző. MHC-hez kötődött peptidek. tumorhoz asszociált antigének (TAA) - embrionális gének termékei- mennyiségi növekedés 50x- 100x. Tumor antigének izolálása: cDNS könyvtárból - CTL segítségével, tumor sejtek felszínéről savas elucióval.")
28
Citotoxikus T sejt klónok előállítása:
in vitro: melanoma sejt tenyészet (tumorból) + limfociták (betegből) Tumor specifikus CTL Tumor cDNS könyvtár transzfektálás MHCI+ célsejtekbe : Tumor antigént bemutató sejtek Együtt tenyésztés, amelyik sejt elpusztult az tartalmazta a megfelelő tumor peptidet Izolálás, szekvenálás tumor ag.
 + limfociták (betegből) Tumor specifikus CTL. Tumor cDNS könyvtár transzfektálás MHCI+ célsejtekbe : Tumor antigént bemutató sejtek. Együtt tenyésztés, amelyik sejt elpusztult az tartalmazta a megfelelő tumor peptidet. Izolálás, szekvenálás tumor ag.")
29
Természetes módon processzált tumor peptidek előállításának módjai
Peptid bejuttatása a sejtbe: Liposzóma (MHCII) Kiméra baktérium (MHCII) Vírus kiméra –minigen (MHCI) Fig. 1. Methods of purification of naturally processed tumor peptides. The steps for the purification of strong extracts (left) and mild extracts (right) are depicted. For mild extracts, a further step of enrichment of material of molecular weight,5000 can be added after the lyophilization.
 Kiméra baktérium (MHCII) Vírus kiméra –minigen (MHCI) Fig. 1. Methods of purification of naturally processed tumor peptides. The steps for the purification. of strong extracts (left) and mild extracts (right) are depicted. For mild extracts, a further step of. enrichment of material of molecular weight,5000 can be added after the lyophilization.")
30
Peptidek terápiás alkalmazása
Szintetikus peptidek Természetes peptidek Előny Immunválasz specifitása sokféle ag keveréke Korlátlan mennyiség több betegnél Nagy tisztaság CD4 CD8 T akt. Kevés tumor sejten ellenőrizhető CD4, CD8 T aktiválás Hátrány Tumorspecifitásra nincs bizonyíték, sok tumor sejt kell Néhány betegnél, alacsony koncentráció Mutáns sejtek megszökhetnek, autoimmunitás kialakulhat Autoimmunitás kialakulhat

31
Therapeutic cancer vaccines
Pramod K Srivastava The immunological bases of current approaches to therapeutic cancer vaccination (or ‘vacci-treatment’) have been established for a decade or longer. The new developments lie mostly in the lessons learnt from clinical testing of these approaches. Three lessons are particularly worthy of note. First, recently completed randomized Phase 3 trials suggest that vacci-treatment with autologous dendritic cells expressing prostatic acid phosphatase (for prostate cancer) or with autologous tumor-derived heat shock protein (gp96)–peptide complexes show promise in enhancing survival of cancer patients. These two approaches are undergoing further randomized clinical testing. Second, immunological monitoring of many clinical trials has failed to identify a surrogate marker for clinical outcomes. Finally, an increasing volume of literature suggests that protective immunity to human cancers is elicited by the mutated antigenic repertoire unique to each cancer.
 have been. established for a decade or longer. The new developments lie. mostly in the lessons learnt from clinical testing of these. approaches. Three lessons are particularly worthy of note. First, recently completed randomized Phase 3 trials suggest. that vacci-treatment with autologous dendritic cells expressing. prostatic acid phosphatase (for prostate cancer) or with. autologous tumor-derived heat shock protein (gp96)–peptide. complexes show promise in enhancing survival of cancer. patients. These two approaches are undergoing further. randomized clinical testing. Second, immunological. monitoring of many clinical trials has failed to identify a. surrogate marker for clinical outcomes. Finally, an increasing. volume of literature suggests that protective immunity to. human cancers is elicited by the mutated antigenic repertoire. unique to each cancer.")
32
Stratégiák a kísérletes tumor immunterápiára
Fig. 1. Current strategies in experimental immunotherapy of tumors. Tumor-specific mAbs can mediate cytolysis either by engaging NK cells via Fc receptors (ADCC), or by complement activation. To reduce immunogenicity of xenogeneic antibodies, constant regions are humanized by recombinant technology. Bispecific antibody constructs are designed to bring immune effector cells into contact with tumor cells and to simultaneously stimulate their cytotoxic activity. Examples include antibodies that recognize a tumor surface antigen and CD16 to activate NK cells, or CD3 to activate T cells. Recombinant fusion products of antitumor antibodies and cytokines can concentrate immune effector functions at the tumor site. CTLs can be activated against tumor antigens by tumor cells rendered immunogenic by expression of either costimulatory molecules such as CD80/86 or cytokines. Effective stimulation of tumor-specific CTLs is achieved by dendritic cells (DCs) presenting major histocompatibility complex class I-bound tumor peptides. DCs are either directly loaded with peptides or exposed to tumor-cell lysates, tumor proteins or DNA. Intracellular synthesis and processing of tumor proteins in DCs is achieved by transfection of cDNA in an expression vector. Abbreviations: ADCC, antibody-mediated cellular cytotoxicity; CTL, cytotoxic T lymphocyte; IL-2, interleukin 2; mAb, monoclonal antibody; NK, natural killer.
, or by complement activation. To reduce immunogenicity of xenogeneic antibodies, constant regions are humanized by recombinant technology. Bispecific antibody constructs are designed to bring immune effector cells into contact with tumor cells and to simultaneously stimulate their cytotoxic activity. Examples include antibodies that recognize a tumor surface antigen and CD16 to activate NK cells, or CD3 to activate T cells. Recombinant fusion products of antitumor antibodies and cytokines can concentrate immune effector functions at the tumor site. CTLs can be activated against tumor antigens by tumor cells rendered immunogenic by expression of either costimulatory molecules such as CD80/86 or cytokines. Effective stimulation of tumor-specific CTLs is achieved by dendritic cells (DCs) presenting major histocompatibility complex class I-bound tumor peptides. DCs are either directly loaded with peptides or exposed to tumor-cell lysates, tumor proteins or DNA. Intracellular synthesis and processing of tumor proteins in DCs is achieved by transfection of cDNA in an expression vector. Abbreviations: ADCC, antibody-mediated cellular cytotoxicity; CTL, cytotoxic T lymphocyte; IL-2, interleukin 2; mAb, monoclonal antibody; NK, natural killer.")
33
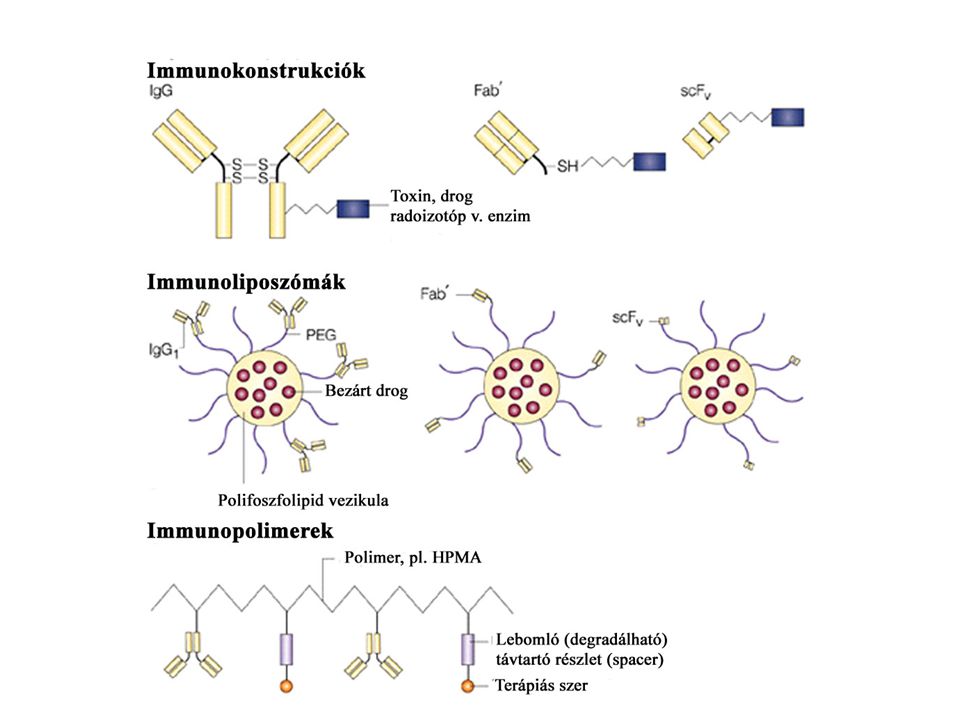
Ellenanyag terápiák

35
Passzív immunterápia:
Passzív immunterápia: Sejtek vagy ellenanyagok közvetítésével Sejtes adaptív terápia: LAK sejtek, TIL - ex vivo kezelt sejtek visszaadása Ellenanyagok. : tumor specifikus immunotoxin: ricin- célzott-dózis alcsonyabb toxin bejut tumorsejtbe koktél ellenanyagok, (antigén vesztés miatt) B lymphoma: CD19, CD20, CD30 specifikus ellenanyagok anti-idiotípus ellenanyagok - ADCC humanizált elenanyagok, egyláncú ellenanyagok heterokonjugátumok: tumor és CTL specifikus - összekapcsol
 B lymphoma: CD19, CD20, CD30 specifikus ellenanyagok. anti-idiotípus ellenanyagok - ADCC. humanizált elenanyagok, egyláncú ellenanyagok. heterokonjugátumok: tumor és CTL specifikus - összekapcsol.")
36
Tumor - ellenanyag terápia
Monoklonális ellenanyagok Előnyök: specifitás, (tumor specifikus (?) ill. tumor-asszociált antigének) homogenitás, affinitás, mellékhatások csökkenése Hátrányok: nagy molekula tömeg - kedvezőtlen biodistribúció, immunogenitás Alkalmazás: ellenanyag (mechanizmus?) immunkonjugátumok: „mágikus golyó”- ellenanyag +radioaktív izotóp, toxin, drog „pro-drog” bi-specifikus ellenanyagok rekombináns ellenanyagok
 ill. tumor-asszociált antigének) homogenitás, affinitás, mellékhatások csökkenése. Hátrányok: nagy molekula tömeg - kedvezőtlen biodistribúció, immunogenitás. Alkalmazás: ellenanyag (mechanizmus ) immunkonjugátumok: „mágikus golyó - ellenanyag +radioaktív izotóp, toxin, drog. „pro-drog bi-specifikus ellenanyagok. rekombináns ellenanyagok.")
37
Monoklonális ellenanyag -drog konjugátumok
Drog: calicheamicin (kétszálú törés DNS-en), doxorubicin - anti- tumor antibiotikumok szelektív - szisztémás toxicitást csökkenti CD33+calicheamicin - AML (akut myeloid leukémia) 20% -nál BR96-DOX - colon carcinoma, tüdőrák, mellrák modell - ember? „minimal” betegség esetén használható Radioaktív jelöléssel ellátott ellenanyagok Radioimmunoterápia (RIT) hosszútávú teljes remisszió gyakori: Non-Hodgkin lymphoma, CLL ellenanyagok: anti-idiotípus CD20, CD22 HLA- DR10
, doxorubicin - anti- tumor antibiotikumok. szelektív - szisztémás toxicitást csökkenti. CD33+calicheamicin - AML (akut myeloid leukémia) 20% -nál. BR96-DOX - colon carcinoma, tüdőrák, mellrák modell - ember „minimal betegség esetén használható. Radioaktív jelöléssel ellátott ellenanyagok. Radioimmunoterápia (RIT) hosszútávú teljes remisszió gyakori: Non-Hodgkin lymphoma, CLL. ellenanyagok: anti-idiotípus. CD20, CD22. HLA- DR10.")
38
B sejtes limfóma kezelése idiotípus-specifikus monoklonális ellenanyaggal
Szelektív kötődés limfóma sejtekhez Komplement- közvetített tumor sejt lízis

39
„Signaling” ellenanyagok
membrán receptorok keresztkötése - szignál intracelluláris jelátadás válasz: apoptózis anti-CD20 (Rituximab) CD20: B sejteken, Ca2+ csatorna, Non-Hodgkin lymphoma: 50% Her2 (neu) (Herceptin) Her-2 protoonkogén terméke, EGFR család mellrák: 14% anti-CD52: Campath-1H CLL, T prolimfocitás leukémia Mab kezelés: nem szünteti meg a tumort - alvó állapot szignaling megváltozik keresztkötés- hipercross-linking Solid tumor: anti-Her-2: receptort gátolja és down-regulálja tirozin kináz jel növekedés gátlás: CDK inhibitor
 CD20: B sejteken, Ca2+ csatorna, Non-Hodgkin lymphoma: 50% Her2 (neu) (Herceptin) Her-2 protoonkogén terméke, EGFR család. mellrák: 14% anti-CD52: Campath-1H. CLL, T prolimfocitás leukémia. Mab kezelés: nem szünteti meg a tumort - alvó állapot. szignaling megváltozik. keresztkötés- hipercross-linking. Solid tumor: anti-Her-2: receptort gátolja és down-regulálja. tirozin kináz jel növekedés gátlás: CDK inhibitor.")
40
Kombinációs terápia: DNA sérülést okozó kemoterápia, sugárterápia hatékonyságát növeli, mellékhatások nélkül Szignál a tumor sejtben Szignál a gazda szervezet immmunrendszerében : CD40, Fas közvetített apoptózis - gátolt a tumor sejtben ellenhatás: effektor sejt apoptózisa - immunterápia: FasL expresszió fokozás tumor sejten

41
”Antibody directed enzyme-prodrog” terápia (ADEPT)
ellenanyag (carciniembrionális antigénre specifikus) F(ab’)2, humanizált, + enzim (pl. alkalikus foszfatáz, aminopeptidáz) konjugátum - anti-enzim ellenanyag (IgG) - enzimet inaktiválja, IC kiürül -pro-drog: nem toxikus, enzimtől függő átalakulás, tumorban lokalizáltan, Előnyök: sok, kis méretű molekula - könnyü penetráció rövid életidő Metoxipolietilén glikol (MPEG) - directed enzyme/prodrog terápia Biokompatibilis, szintetikus polimer - tumorban felgyülemlik Immunogenitás csökken- ismételten használható Immunszupresszió nem szükséges
 F(ab’)2, humanizált, + enzim (pl. alkalikus foszfatáz, aminopeptidáz) konjugátum. - anti-enzim ellenanyag (IgG) - enzimet inaktiválja, IC kiürül. -pro-drog: nem toxikus, enzimtől függő átalakulás, tumorban lokalizáltan, Előnyök: sok, kis méretű molekula - könnyü penetráció. rövid életidő Metoxipolietilén glikol (MPEG) - directed enzyme/prodrog terápia. Biokompatibilis, szintetikus polimer - tumorban felgyülemlik. Immunogenitás csökken- ismételten használható. Immunszupresszió nem szükséges.")
42
Enzim - Pro-drog terápia

43
Adoptív sejt-transzfer terápia: limfocitákat depletáló kemoterápia után
Box 1 | Adoptive-cell-transfer therapy for patients with cancer Adoptive-cell-transfer therapy provides three opportunities for immune manipulation, which are not readily achieved with other immunotherapeutic approaches. First, highly active, tumour-reactive lymphocyte cultures with optimal characteristics can be selected. For patients with melanoma, the generation of tumour-infiltrating lymphocyte (TIL) cultures was optimized to produce highly avid, tumour-antigen-reactive cells that secreted high levels of interferon- (IFN- ). This was accomplished by establishing several, independent TIL lines and assaying each line for recognition of a panel of melanoma cells. The most active TIL lines were identified by enzyme-linked immunosorbent assay (ELISA) and were selected for further expansion (see a). Second, lymphocyte cultures can be rapidly expanded ex vivo, circumventing normal immune-regulatory mechanisms and obviating a potentially suppressive tumour environment. For treatment of patients with melanoma, the highly selected TIL cultures were rapidly expanded using anti-CD3 antibody, exogenously supplied interleukin 2 and irradiated allogeneic peripheral blood mononuclear 'feeder' cells (see b). Third, a patient can receive systemic immunosuppression without compromising the antitumour lymphocytes that are being cultured ex vivo. Patients with melanoma received a non-myeloablative but lymphodepleting chemotherapy regimen (see c). These three immune manipulations can be theoretically applied to treat patients with any tumour histology, and they have been successfully combined for the treatment of patients with metastatic melanoma. Melanoma metasztázis kezelésére
 cultures was optimized to produce highly avid, tumour-antigen-reactive cells that secreted high levels of interferon- (IFN- ). This was accomplished by establishing several, independent TIL lines and assaying each line for recognition of a panel of melanoma cells. The most active TIL lines were identified by enzyme-linked immunosorbent assay (ELISA) and were selected for further expansion (see a). Second, lymphocyte cultures can be rapidly expanded ex vivo, circumventing normal immune-regulatory mechanisms and obviating a potentially suppressive tumour environment. For treatment of patients with melanoma, the highly selected TIL cultures were rapidly expanded using anti-CD3 antibody, exogenously supplied interleukin 2 and irradiated allogeneic peripheral blood mononuclear feeder cells (see b). Third, a patient can receive systemic immunosuppression without compromising the antitumour lymphocytes that are being cultured ex vivo. Patients with melanoma received a non-myeloablative but lymphodepleting chemotherapy regimen (see c). These three immune manipulations can be theoretically applied to treat patients with any tumour histology, and they have been successfully combined for the treatment of patients with metastatic melanoma. Melanoma metasztázis kezelésére.")
44
Vaccines to protect against HPV infection and the subsequent risk of cervical cancer have recently become available, based on virus-like particle (VLP) technology. VLPs are produced by expressing the capsid proteins of the virus using recombinant DNA technology. When expressed in eukaryotic cells, the L1 major capsid protein self-assembles into 360-mer particles that physically and immunologically resemble the native virion. These highly immunogenic particles can, when administered as a vaccine with alum based adjuvants, protect not only against infection with the HPV types incorporated in the vaccine and some cross-reactive HPV types but also against the consequent premalignant disease Protection is durable over at least five years, and the vaccine appears to protect nearly 100% of immunised subjects. Human Papilloma Virus Az összes tumoros megbetegedés 5-10%-a HPV fertőzés következménye. A méhnyakrák 100%-ban HPV fertőzés következménye!

45
Human papilloma virus

47
A tumornövekedéssel párhuzamosan kifejlődő anti-tumor válasz modellje
A tumorsejtek aktívan elnyomják a kialakuló tumor-specifikus immunválaszt Figure 1 | A probable model of the evolution and fate of antitumour immune responses that develop coincidently with tumour growth. a | Tumours develop over a long period of time through a process of accumulation of many mutations. While the tumour is small and does not present a significant danger to the integrity of the organ of origin, the immune system remains ignorant of its presence. Dendritic cells (DCs) in the surrounding tissue are not activated and as a result T and B cells in the lymph node remain in a resting state. b | When the tumour becomes larger, heterogeneous and ultimately malignant, damage to the normal tissue and products made by the tumour cells alert the immune system mainly through the activation of resident DCs. Activated DCs that have taken up products derived from damaged normal tissues and tumours, traffic to the draining lymph node in which they begin to present these products as antigens to naive T and B cells. The extent of DC activation determines the extent of lymphocyte stimulation. This in turn is regulated by many factors that determine the immune competence of the patient, including age. c | Tumour-specific T cells, antibodies and activated DCs reach the tumour site and attempt to destroy the tumour. They are only partially successful owing to an already large tumour size and marked tumour heterogeneity that allows the tumour to evade many immune effector mechanisms. d | The tumour that has evaded the initial immune response continues to grow, disseminate and actively suppress local, as well as systemic, immunity illustrated by the presence at the tumour site of DCs, T and B cells that are not activated and do not exert their respective functions.
 in the surrounding tissue are not activated and as a result T and B cells in the lymph node remain in a resting state. b | When the tumour becomes larger, heterogeneous and ultimately malignant, damage to the normal tissue and products made by the tumour cells alert the immune system mainly through the activation of resident DCs. Activated DCs that have taken up products derived from damaged normal tissues and tumours, traffic to the draining lymph node in which they begin to present these products as antigens to naive T and B cells. The extent of DC activation determines the extent of lymphocyte stimulation. This in turn is regulated by many factors that determine the immune competence of the patient, including age. c | Tumour-specific T cells, antibodies and activated DCs reach the tumour site and attempt to destroy the tumour. They are only partially successful owing to an already large tumour size and marked tumour heterogeneity that allows the tumour to evade many immune effector mechanisms. d | The tumour that has evaded the initial immune response continues to grow, disseminate and actively suppress local, as well as systemic, immunity illustrated by the presence at the tumour site of DCs, T and B cells that are not activated and do not exert their respective functions.")
48
Az anti-tumor válasz manipulációja terápiás célú vakcináció által, a tumor kialakulása után
immunogén Figure 2 | Manipulation of antitumour immune responses by therapeutic vaccination. a | Therapeutic vaccines are administered after the tumour is diagnosed, at the time of interactions between the tumour and the immune system that correspond to parts c and d in Fig. 1. In the most optimal clinical setting, therapeutic vaccines intend to boost immunity against minimal residual disease and prevent the outgrowth of metastases shown in parts b and c. A vaccine based on autologous tumour or defined tumour antigens is administered in an immunostimulatory preparation (with adjuvant) that can activate Langerhans cells — dendritic cells (DCs) that reside in the epidermis. Activated Langerhans cells take up the tumour antigens and traffic to the draining lymph node in which they present antigens to T cells. B cells are also activated and the expected outcome is clonal expansion of tumour-specific T cells and the production of tumour-specific antibodies. b | Tumour-specific T cells migrate to the sites of tumour metastases where they attempt to kill tumour cells that express antigens contained in the vaccine. Their function is compromised by the immunosuppressive tumour microenvironement, which affects their function and leads to their death. Furthermore, tumour heterogeneity has been established over time. Some tumour cells have lost expression of antigens that are targeted by the immune response and others have become resistant to immune effector mechanisms. This allows many of the cells to evade the immune attack. c | Metastases that continue to grow are composed of tumour cells that lack antigens recognized by T cells and antibodies or are otherwise resistant to immune destruction.
 that can activate Langerhans cells — dendritic cells (DCs) that reside in the epidermis. Activated Langerhans cells take up the tumour antigens and traffic to the draining lymph node in which they present antigens to T cells. B cells are also activated and the expected outcome is clonal expansion of tumour-specific T cells and the production of tumour-specific antibodies. b | Tumour-specific T cells migrate to the sites of tumour metastases where they attempt to kill tumour cells that express antigens contained in the vaccine. Their function is compromised by the immunosuppressive tumour microenvironement, which affects their function and leads to their death. Furthermore, tumour heterogeneity has been established over time. Some tumour cells have lost expression of antigens that are targeted by the immune response and others have become resistant to immune effector mechanisms. This allows many of the cells to evade the immune attack. c | Metastases that continue to grow are composed of tumour cells that lack antigens recognized by T cells and antibodies or are otherwise resistant to immune destruction.")
49
Az anti-tumor válasz manipulációja megelőző célú vakcináció által
Figure 3 | Manipulation of antitumour immune responses by prophylactic vaccination. a | Prophylactic vaccines would be administered before the occurrence of tumours to individuals who are at high risk for developing tumours or have been diagnosed with premalignant changes in target tissues. A vaccine based on antigen/s that are expected to be expressed by the anticipated tumour is administered in an immunostimulatory preparation (with adjuvant) that can activate Langerhans cells — dendritic cells (DCs) that reside in the epidermis. Activated Langerhans cells take up the tumour antigens and traffic to the draining lymph node in which they present antigens to T cells. B cells are also activated and the expected outcome is clonal expansion of tumour-specific T cells and the production of tumour-specific antibodies. This clonal expansion of effector cells is followed in time by the generation of a pool of memory cells that are specific for the tumour antigen/s. b | If a tumour begins to grow sometime in the future, tumour antigens that reach the draining lymph node will reactivate tumour-specific memory cells and elicit a swift secondary immune response. This response will be characterized by large numbers of effector T cells, high titre of antibodies and continuous activation of DCs at the tumour site, for continuous processing and presentation of tumour antigens and further amplification of the immune response. c | The incipient tumour has not been allowed to grow large and heterogeneous and is easily eliminated by the prepared immune response. Moreover, the memory compartment is further expanded by this tumour-mediated boost. Tumor-specifikus memóriasejtek reaktiválása
 that can activate Langerhans cells — dendritic cells (DCs) that reside in the epidermis. Activated Langerhans cells take up the tumour antigens and traffic to the draining lymph node in which they present antigens to T cells. B cells are also activated and the expected outcome is clonal expansion of tumour-specific T cells and the production of tumour-specific antibodies. This clonal expansion of effector cells is followed in time by the generation of a pool of memory cells that are specific for the tumour antigen/s. b | If a tumour begins to grow sometime in the future, tumour antigens that reach the draining lymph node will reactivate tumour-specific memory cells and elicit a swift secondary immune response. This response will be characterized by large numbers of effector T cells, high titre of antibodies and continuous activation of DCs at the tumour site, for continuous processing and presentation of tumour antigens and further amplification of the immune response. c | The incipient tumour has not been allowed to grow large and heterogeneous and is easily eliminated by the prepared immune response. Moreover, the memory compartment is further expanded by this tumour-mediated boost. Tumor-specifikus memóriasejtek reaktiválása.")
50
?FcgRIIb gátlás, vagy IgG Fc megváltoztatása mellett IC adása DC-nek?
Inducing Tumor Immunity through the Selective Engagement of Activating Fc Receptors on Dendritic Cells A gátló FcgRIIb eltávolítása a dendritikus sejtekről fokozza azok érését és anti-tumor hatását Tumor elleni vakcina előállítása: ?FcgRIIb gátlás, vagy IgG Fc megváltoztatása mellett IC adása DC-nek? Figure 3. Removal of inhibitory Fc receptor signaling on DCs enhances their ability to protect against tumors. Day 6 DC cultures derived from bone marrows of either WT or FcRIIB/ mice (C57BL/6 background) were incubated for 6 h with 50 g/ml of ICs made of OVA and anti-OVA rabbit IgG. DCs were washed in PBS and injected in the footpads of naive syngeneic C57BL/6 mice. 2 wk after this single immunization, mice were challenged subcutaneously with a variant of the melanoma B16 tumor line that expresses OVA as a neo-antigen (MO4) (references 28–30) ( cells per mouse). Tumor growth was monitored three times a week and data from three independent experiments are shown. (A) Scheme for DC immunization and tumor challenge. (B) Tumor growth curves for mice immunized with OVA–IC-pulsed DCs and challenged with B16-OVA (mice showing palpable tumors). While WT DCs and FcRIIb/ DCs are statistically different (P ), naive and WT DCs are not (P 0.80). (C) Tumor appearance for mice immunized with OVA–ICs and challenged with B16-OVA. Mice were considered positive when palpable tumors were detected. While naive and FcRIIb/ DCs are statistically different (P ), naive and WT-DC are not (P ). Alexis M. Kalergis and Jeffrey V. Ravetch
 were incubated for 6 h with 50 g/ml of ICs made of OVA and anti-OVA rabbit. IgG. DCs were washed in PBS and injected in the footpads of naive syngeneic C57BL/6 mice. 2 wk after this single immunization, mice were challenged subcutaneously with a variant of the melanoma B16 tumor line that expresses OVA as a neo-antigen (MO4) (references 28–30) (5 105 cells per mouse). Tumor growth was monitored three times a week and data from three independent experiments are shown. (A) Scheme for DC immunization and tumor challenge. (B) Tumor growth curves for mice immunized with OVA–IC-pulsed DCs and challenged with. B16-OVA (mice showing palpable tumors). While WT DCs and FcRIIb/ DCs are statistically different (P 0.002), naive and WT DCs are not (P 0.80). (C) Tumor appearance for mice immunized with OVA–ICs and challenged with B16-OVA. Mice were considered positive when palpable. tumors were detected. While naive and FcRIIb/ DCs are statistically different (P ), naive and WT-DC are not (P 0.079). Alexis M. Kalergis and Jeffrey V. Ravetch.")
Hasonló előadás


 daganatkeltő.>")













>")
>")



