Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
The a-amino group Transamination Exception Pro Hyp Thr Lys

2
Catabolism of carbonic skeleton of amino acids
Medical importance: Disease - low frequency Mental retardation Prenatal diagnosis Postnatal diagnosis - treatment

3
Catabolism of carbon skeleton
Amphybolic intermedier Glucoplastic 13 ketoplastic 1 Gluco- and ketoplastic 5 Ala, Arg, Asp, Cys, Glu, Gly, His, Hyp, Met, Pro, Ser, Thr, Val Leu Ile, Lys, Phe, Trp, Tyr

4
Ala, Cys, Gly, Hyp, Ser, Thr Arg, His Gln, Pro Ile, Met, Val Tyr, Phe
l-Glutamate Ile, Leu,Trp Pyruvate a-Ketoglutarate Ile, Met, Val Citrate Acetyl-CoA Succinyl-CoA Acetoacetyl-CoA Citrat cyclus Leu, Lys, Phe, Trp, Tyr Oxalacetat Fumarat Tyr, Phe l-Aspartat l-Asn

5
Asn, Asp - Oxaloacetate Asparaginase Transaminase

6
Ala, Cys, Gly, Hyp, Ser, Thr Arg, His Gln, Pro Ile, Met, Val Tyr, Phe
l-Glutamate Ile, Leu,Trp Pyruvate a-Ketoglutarate Ile, Met, Val Citrate Acetyl-CoA Succinyl-CoA Acetoacetyl-CoA Citrat cyclus Leu, Lys, Phe, Trp, Tyr Oxalacetat Fumarat Tyr, Phe l-Aspartat l-Asn

7
Gln & glu a-ketoglutarate
Glutaminase Transaminase

8
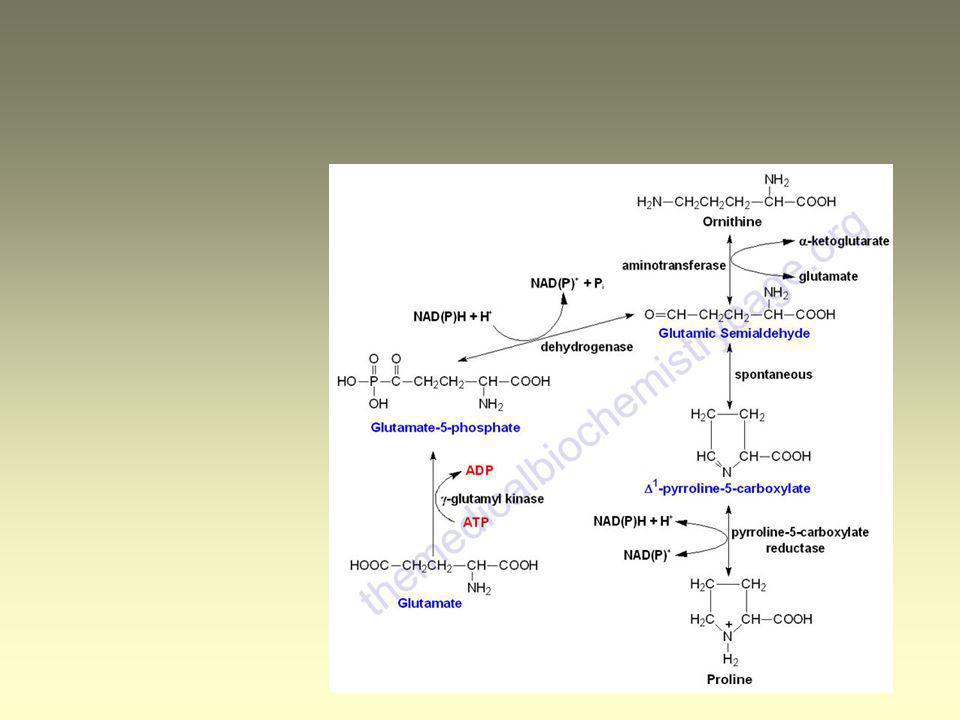
Pro - a-ketoglutarate Proline Arginine L-glutamat-g-semialdehyd

10
Pro - a-ketoglutarate (1)
")
11
Pro - a-ketoglutarate (2)
")
12
Pro - a-ketoglutarate 2 autosomal recessive hyperprolinemia
Hyperprolinaemia I: prolin-dehydrogense (Hz –symptoms of hyperprolinaemia) Hyperprolinaemia II: Glutamate-g-semialdehyd dehydrogenase (hyperhydroxypolinaemia, Hz absence of symptoms of hyperprolinaemia)
 Hyperprolinaemia II: Glutamate-g-semialdehyd dehydrogenase (hyperhydroxypolinaemia, Hz absence of symptoms of hyperprolinaemia)")
13
Arg & ornitin - a-ketoglutarate

14
Arg & ornitin - a-ketoglutarate
The defect of ornitin d-aminotransferase results in: [ornitin] is enhanced, blindness. Hyperornitinaemia – hyperammonaemia syndrom: [ornitin]plazma is enhanced. Lowered mitochondrial transport.

15
L-His - a-ketoglutarate
Histidase Histidinaemia Urokanate Urokanase Urokaninic acidaemia 4-imidazolon-5-propionate ~ dehidrogenase F(iglu) Glutamate forminino transferase Folic acid deficiency / test L-Glu transaminase a-ketoglutarate
 Glutamate forminino transferase. Folic acid deficiency / test. L-Glu. transaminase. a-ketoglutarate.")
16
L-His - a-ketoglutarate

17
L-His - a-ketoglutarate
Histininaemia Histidase enzyme defect: 1:11500 [His]blood, urine is elevated Typical impediment in speech, benign syndrome Urokaninic aciduria Autosomal recessive inheritance [His]urine is elevated benign syndrome

18
Ala, Cys, Gly, Hyp, Ser, Thr Arg, His Gln, Pro Ile, Met, Val Tyr, Phe
l-Glutamate Ile, Leu,Trp Pyruvate a-Ketoglutarate Ile, Met, Val Citrate Acetyl-CoA Succinyl-CoA Acetoacetyl-CoA Citrat cyclus Leu, Lys, Phe, Trp, Tyr Oxalacetat Fumarat Tyr, Phe l-Aspartat l-Asn

19
Piruvat-dehydrogenase
Amino acids - pyruvate L-Threonin L-Glycine Cystine L-Serin Pyruvat L-Alanine L-Cystein Piruvat-dehydrogenase Acetyl-CoA

20
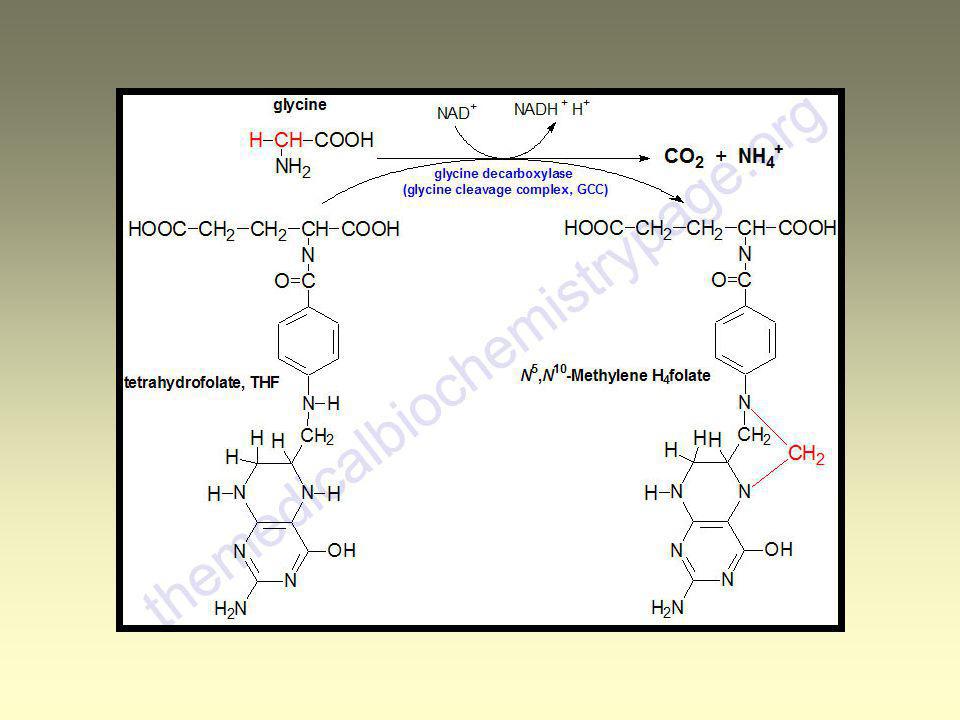
L-Glycin Synthesis of glutathion, creatin, purine skeleton, conjugated bile acids, hem Glucoplastic aminoacid Catabolism of Gly: Glycin – Serin – Pyruvate – Acetyl-CoA Glycine cleavage – Glycine synthase complex

21
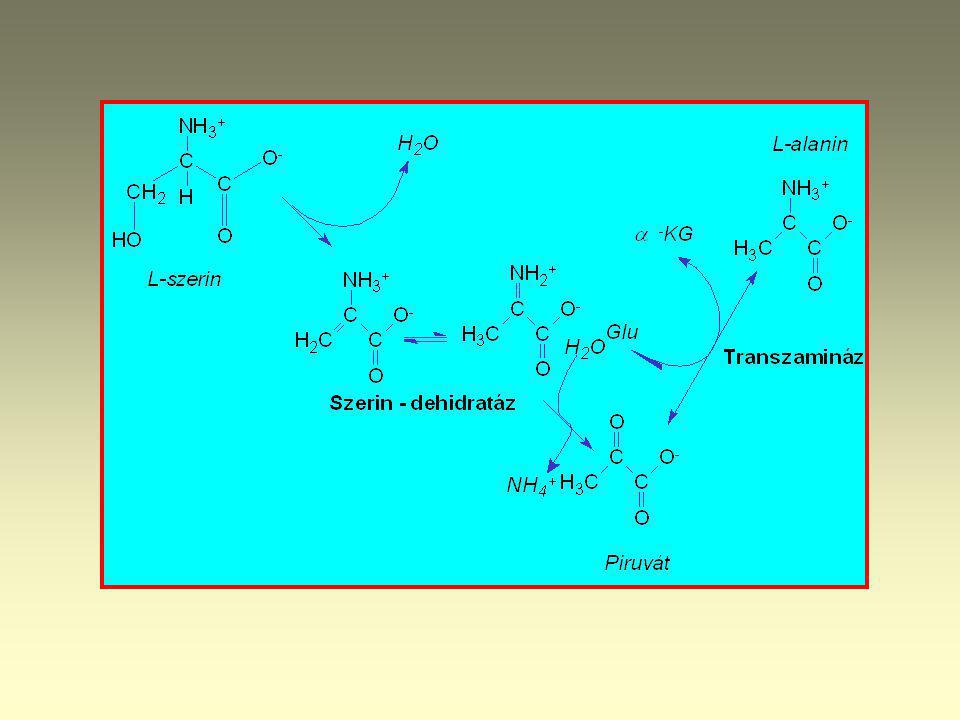
L-Glycin Serin hydroxymethyl transferase

22
Cleavage of Gly by mitochondrial glycine synthase
complex

24
L-Glycin Glycinuria: Primary hyperoxaluria 0,6 – 1 g glycin/day
Oxalate-typ neprolyth Defect in tubular reabsorption of kidneys Primary hyperoxaluria Glycin deamination – glyoxilate, oxalate Ca-oxalate type neprolyths

25
Amino acids - pyruvate L-Threonin L-Glycine Cystine L-Serin Piruvate
L-Alanin L-Cysteine Acetyl-CoA

27
Amino acids - pyruvate L-Threonin L-Glycin Cystine L-Serin Piruvat
L-Alanin L-Cysteine Acetyl-CoA

28
Cystin – cystein conversion

29
Cystein – pyruvate conversion: 2 ways
Direct oxydative: cystein sulfinate Transamination: 3-merkaptopyruvate „activ” sulfate formation (3’-phosphoadenosine-5’-phosphosulfate) Glutathion syntesis
 Glutathion syntesis.")
30
Cystein – pyruvate conversion: (i) direct oxydative way
Cystein dioxygenase Fe2+ NAD(P)H Desulfinase / spontan reaction Taurine
H. Desulfinase / spontan reaction. Taurine.")
31
Cystein – pyruvate conversion : (ii) transamination
3-merkapto-lactate – in human urin ~ + cysteinnel disulfid – in urine [merkaptolactat – cystein]urine merkaptolactate – in cystein disulfid uria

32
3'-phosphoadenosine-5'-phosphosulfate, (PAPS).
.")
33
A kén tartalmú aminosavak lebontásának rendellenességei
Cisztinuria (cisztin – lizin uria) Cisztinózis Homociszteinuriák
 Cisztinózis. Homociszteinuriák.")
34
Amino acids - pyruvate L-Threonin L-Glycin CO2 + NH4+ Cystine L-Serin
Piruvat L-Alanin L-Cysteine Acetyl-CoA

35
Treonin: treonin aldolase è two ways
Acetaldehyde threonin aldolase Glycine è Methylen H4 folate +CO2+NH4+ L-serine Piruvate

36
Glyoxylate is formed from 4-hydroxyprolin képződik
4-hidroxi-prolin Hidroxiprolin dehidrogenáz L-D1-Pirroline-3-hidroxi-5-karboxilát Nem enzimatikus g-hydoxi-l-glutamát-g-szemialdehid dehidrogenáz Eritro-g-hidroxi-l-glutamát transzamináz a-keto-g-hidroxiglutarát aldoláz Glioxilát + piruvát

37
4-hydroxyprolin piruvate & glyoxylate

38
4-hydroxyprolin piruvate & glyoxylate
Hyperhydroxyprolinaemia: Hydroxyproline dehydrogenase [4-hydroxyproline]plasma Autosomal resessive trait Glutamate-g-semialdehyde dehydrogenase L-D1-Pyrroline-3-hydroxi-5-carboxilate Lack of hyperprolinaemia

Hasonló előadás











 P(b) Alanine 142 83 Arginine 98 93 Aspartic Acid 101 54 Asparagine 67 89.>")







