Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
Felnőttkorban felismert hereditaer nephropathiák
Dr. Mátyus János egyetemi docens DEOEC Belgyógyászati Intézet, I. sz. Belgyógyászati Klinika, Nephrologiai Tanszék, Debrecen

2
Felnőttkorban felismert hereditaer nephropathiák
Mátyus János DE OEC I. Belkl. XV.DNN

3
Nephrológusok érdeklődése csökken a herediter nephropathiák iránt
„Gyermek nephrológusok dolga” „Felnőttkorban nincs jelentőségük (kiv. ADPKD)” „Nincs oki kezelési lehetőség (kiv. Fabry)” „Drága v. nem hozzáférhető genetikai diagnózis” „Gyakori kórképek genetikai vonatkozásait kutassuk” pl. ACE, Dvit. rec. polimorf. Felismerésüket nehezíti negatív családi anamnézis AR v. X öröklés, csökkenő gyermekszám AD: új mutáció (pl. ADPKD 10%, X-L Alport 15%) változatos penetrancia és megjelenés ismeretek és idő hiánya
 „Nincs oki kezelési lehetőség (kiv. Fabry) „Drága v. nem hozzáférhető genetikai diagnózis „Gyakori kórképek genetikai vonatkozásait kutassuk pl. ACE, Dvit. rec. polimorf. Felismerésüket nehezíti. negatív családi anamnézis. AR v. X öröklés, csökkenő gyermekszám. AD: új mutáció (pl. ADPKD 10%, X-L Alport 15%) változatos penetrancia és megjelenés. ismeretek és idő hiánya.")
4
H.D.Anett sz.1982. Anamnesis: cystitis 1x, egyéb betegsége nem volt, dohányzik bo. szúró mellkasi fájd. tüdőgond.: pneumonia, ab-ra nem javul belgyógy.: embolia pulm. (izotóppal), avtgi thromb. nincs OAC szedés 1 éve, proteinuria>4g/n, mikr.haematuria vesebiopsia: habos tub. hám (glom. nincs) 1mg/kg po. steroid 12 hétig hatástalan : DEOEC rebiopsia: Alport sy szemészet, hallásvizsg, szülők: eltérés nincs 2 hónapig: 4mg/kg CyA+24mg MP: hatástalan 5 hónapig: 1,5mg/kg Cytoxan+mn. 16mg MP: hatástalan
, avtgi thromb. nincs OAC szedés 1 éve, proteinuria>4g/n, mikr.haematuria vesebiopsia: habos tub. hám (glom. nincs) 1mg/kg po. steroid 12 hétig hatástalan : DEOEC rebiopsia: Alport sy. szemészet, hallásvizsg, szülők: eltérés nincs. 2 hónapig: 4mg/kg CyA+24mg MP: hatástalan. 5 hónapig: 1,5mg/kg Cytoxan+mn. 16mg MP: hatástalan.")
5
Herediter nephropathiák vizsgálata
gyanú felvetése (gondolni kell rá !) gondos családi anamnesis, családfa vizsgálat kivizsgálás (családtagok is), finom jelek ! vesebetegség (u-MA, u-B2m, u-ül., GFR) extrarenalis tünetek keresése (pl. szem, fül) képalkotó - szövettani vizsg. (jó radiol.-pathol.) ismert gén keresése adatbázisokban www3.ncbi.nlm.nih.gov/OMIM genetikai diagnosztika, ha rendelkezésre áll ismeretlen gén keresése, ha nagy a család marker gének, géntérkép, jó laboratórium
 gondos családi anamnesis, családfa vizsgálat. kivizsgálás (családtagok is), finom jelek ! vesebetegség (u-MA, u-B2m, u-ül., GFR) extrarenalis tünetek keresése (pl. szem, fül) képalkotó - szövettani vizsg. (jó radiol.-pathol.) ismert gén keresése adatbázisokban. www3.ncbi.nlm.nih.gov/OMIM. genetikai diagnosztika, ha rendelkezésre áll. ismeretlen gén keresése, ha nagy a család. marker gének, géntérkép, jó laboratórium.")
6
Felnőttkori polycystas vesebetegség (ADPKD)
leggyak. hered.: 1:400-1:1.000, dializáltak 4-8% öröklés: AD (teljes penetrancia, vált. expressio) ADPKD1 16p13.1, kaukázusiak 85%-ában ok, polycystin (sejt-sejt, sejt-matrix kapcsolat) ADPKD2 4q21-23 (lassabb progressio) ADPKD3 ismeretlen gyakori spontán mutáció: 10% génanalysis (PKD1) elérhető, de in-utero nem jav. (psychológiai vonzat, lefolyást nem jelzi!) dg: UH, ha 1.évben cysta: 30% VE 18év előtt 30.év nincs cysta: (10-15%), VE nem lesz
 ADPKD1 16p13.1, kaukázusiak 85%-ában ok, polycystin (sejt-sejt, sejt-matrix kapcsolat) ADPKD2 4q21-23 (lassabb progressio) ADPKD3 ismeretlen. gyakori spontán mutáció: 10% génanalysis (PKD1) elérhető, de in-utero nem jav. (psychológiai vonzat, lefolyást nem jelzi!) dg: UH, ha 1.évben cysta: 30% VE 18év előtt 30.év nincs cysta: (10-15%), VE nem lesz.")
7
ADPKD renális manifesztációi
hypertonia (ált. 30év után kezd) VE-ben 80% ok: cysta nő, ter.: punctio!, ACEI idült veseelégtelenség 60 éves korban 45% húgyúti infectio (pyelonephr., cysta) közel 100% fájdalom (acut-chr.) % acut: cysta bevérzés, obstructio, infectio haematuria (spontán-trauma) 50% ok: cysta ruptura-kő-infectio-veserák ter.: ágy, DDAVP, transfúzió, opus/emb. vesekő: ok: pangás, tart.: húgysav/Ca-oxal %, dg.: IVU/CT, ter.: endourol.,ESWL veserák? dg.: haematu., fájd., dg.:CT/MRI 5%
 VE-ben 80% ok: cysta nő, ter.: punctio!, ACEI. idült veseelégtelenség 60 éves korban 45% húgyúti infectio (pyelonephr., cysta) közel 100% fájdalom (acut-chr.) 60% acut: cysta bevérzés, obstructio, infectio. haematuria (spontán-trauma) 50% ok: cysta ruptura-kő-infectio-veserák ter.: ágy, DDAVP, transfúzió, opus/emb. vesekő: ok: pangás, tart.: húgysav/Ca-oxal. 20%, dg.: IVU/CT, ter.: endourol.,ESWL. veserák dg.: haematu., fájd., dg.:CT/MRI 5%")
8
ADPKD extrarenalis manifesztációi
GI: májcysta 50%: norm. enzimek, ritkán infectio igen ritka: cholangiocc., congenit. májfibr. pancreas cysta 10%, ovarium cy, colon divert.? mitralis prolapsus 25%, szöv.: ritmuszavar, endocarditis, embolisatio agyi aneurysma 5-10% (mortalitas gyakori oka) szöv.: aneurysma rupt., intracerebr. vérzés tü: erős-szokatlan fejfájás, hányinger dg.: szűrés: koponya MRI? tünet esetén: DSA,CT/MRI ter.: idegseb., interv. rad.
 szöv.: aneurysma rupt., intracerebr. vérzés tü: erős-szokatlan fejfájás, hányinger dg.: szűrés: koponya MRI tünet esetén: DSA,CT/MRI ter.: idegseb., interv. rad.")
9
Vesecyták felnőttkorban (ADPKD diff. dg.)
szerzett: szerzett cystas vesebetegség egyszerű cysta pl. Conn sy-ban herediter betegségek cystak vesetumorral sclerosis tuberosa vonHippel-Lindau vesecysták idült tubuloint. nephropathiával medullaris cystas vese1/FJHN MCKD2 nephronophtisis 3.-4.tip. (AR) vesecysták diabetessel
 vesecysták diabetessel.")
10
Szerzett cystás vesebetegség (ACKD)
fogalma: a veseparenchyma cystás átalakulása idült, hosszantartó veseelégtelenségben alapbetegség: nem herediter cystás vesebetegség kivételével bármelyik (?) gyakoriság: döntő a VE hossza és súlyossága ! predialízis: 10-15%, majd évi 5-10% dialízis 3 év: >50%, 10év: >90% független: életkor, nem(ffi?), race (feketék?) dialízis mód (PD-HD), gyógyszerszedés (cyclosporin?)
 gyakoriság: döntő a VE hossza és súlyossága ! predialízis: 10-15%, majd évi 5-10% dialízis 3 év: >50%, 10év: >90% független: életkor, nem(ffi ), race (feketék ) dialízis mód (PD-HD), gyógyszerszedés (cyclosporin )")
11
Örökletes és szerzett cystás vesebetegség elkülönítése
veserák

12
Uromodulin nephropathiák
Korábbi elnevezések: medullaris cystas vese (MCKD2) fam. juvenilis hyperuricemias nephropathia (FJHN) Gyakoriság: nem ismert Öröklés: AD UMOD: 16p12 uromodulin főleg cisztein vesztő mutációk a 4-5. exonban (MCKD2/FJHN) ismeretlen: 1q21 (MCKD1) Uromodulin (Tamm-Horsfall) 30-50mg/n, felszálló Henle cylinderképz., baktérium / kőképződés ell. védelem Pathomech.: diszulfid hidak kialakulása↓ → károsodik a harmadlagos szerkezet → lassul az endopl. ret-ból a Golgiba jutás, retineálódik →tubulus apoptosis
 fam. juvenilis hyperuricemias nephropathia (FJHN) Gyakoriság: nem ismert. Öröklés: AD. UMOD: 16p12 uromodulin főleg cisztein vesztő mutációk a 4-5. exonban (MCKD2/FJHN) ismeretlen: 1q21 (MCKD1) Uromodulin (Tamm-Horsfall) 30-50mg/n, felszálló Henle. cylinderképz., baktérium / kőképződés ell. védelem. Pathomech.: diszulfid hidak kialakulása↓ → károsodik a harmadlagos szerkezet → lassul az endopl. ret-ból a Golgiba jutás, retineálódik →tubulus apoptosis.")
13
FJHN/MCKD2 klinikum Lassú progressziójú tubuloint. nephritis
polyuria, polydipsia nincs hematuria/proteinuria, neg. üledék hypertonia ritka scr↑ 20é, ESRD 40é (homozygotákban gyorsabb) Hyperuricemia, ritkábban köszvény fiúkban már 15-20é, de lehet norm. is (nők) ok: urina húgysav alacsony, FE<6% Dg: fam. előzmény hiányában: genetikai UH: medulláris cysta lehet, sokszor nincs ? vizelet uromodulin cc↓ ? vbiopsia: diffúz tubuloint.fibrosis, nincs hs kristály Diff.dg: egyéb okú TIN kizárandó Kezelés: allopurinol (korán)?, colchicin?? veseTx
 Hyperuricemia, ritkábban köszvény. fiúkban már 15-20é, de lehet norm. is (nők) ok: urina húgysav alacsony, FE<6% Dg: fam. előzmény hiányában: genetikai. UH: medulláris cysta lehet, sokszor nincs. vizelet uromodulin cc↓ vbiopsia: diffúz tubuloint.fibrosis, nincs hs kristály. Diff.dg: egyéb okú TIN kizárandó. Kezelés: allopurinol (korán) , colchicin veseTx.")
14
Nephronophtisis (NPHP)
Gyak.: gyermek-fiatalkori VE 10-15% Genetika: AR, 9 különböző gén NPHP 1-9 nephrocystinek (tub. sejtek, microvillusok) Klin.: polyuria, polydipsia, nincs proteinu-haemat anaemia, növekedés retardáció progr. VE, gyakran cysták, sóvesztés, acidosis Juv. 1.tip. 2q13 ESRD 13é Infant. 2.tip. 9q é Aldol. 3.tip. 3q é 4.tip. 1p é Társul: retinitis pigm.(5.6.) ataxia (6.7.), májfibr.
 Klin.: polyuria, polydipsia, nincs proteinu-haemat. anaemia, növekedés retardáció. progr. VE, gyakran cysták, sóvesztés, acidosis. Juv. 1.tip. 2q13 ESRD 13é. Infant. 2.tip. 9q31 1-3é. Aldol. 3.tip. 3q é. 4.tip. 1p é. Társul: retinitis pigm.(5.6.) ataxia (6.7.), májfibr.")
15
Renal cysts and diabetes sy (RCAD)
Genetika: AD, 17q12 hepatocyta nucleáris faktor-1β (HNF1β) transzkripciós f.: vese-pancreas-máj-genitalia-bél PKHD1 traszkripciót gátol gyakori spontán mutáció 50%-ban deléció (nehéz kimutatni: MLPA) Vesefejlődési zavarok (ez a leggyak. ok?) cysta, cystas dysplasia (lehet antenatalis– nincs 20%) aplasia, hypoplasia, cong. hydronephrosis glomerulocystás vese, oligomeganephronia változatos vesefunkció: normális →ESRD Diabetes: rendszerint 15-30é korban, de sokszor nincs többnyire inzulin kell (MODY5), pancreas atrófia lehet Egyéb: májenzim, epeúti eltérés, uterus rendell.
 transzkripciós f.: vese-pancreas-máj-genitalia-bél PKHD1 traszkripciót gátol. gyakori spontán mutáció. 50%-ban deléció (nehéz kimutatni: MLPA) Vesefejlődési zavarok (ez a leggyak. ok ) cysta, cystas dysplasia (lehet antenatalis– nincs 20%) aplasia, hypoplasia, cong. hydronephrosis. glomerulocystás vese, oligomeganephronia. változatos vesefunkció: normális →ESRD. Diabetes: rendszerint 15-30é korban, de sokszor nincs. többnyire inzulin kell (MODY5), pancreas atrófia lehet. Egyéb: májenzim, epeúti eltérés, uterus rendell.")
16
G.U.Ryffel,

17
Kerecuk NDT 2007;22:259. asympt. GFR:77ml/p j:9,0, b:10,4cm
19é cystitis, PU 0,8g/n 21é „min.ch.NP” 8,8cm, GFR:70 31é PU, solit.vese 37é FSGS 53é veseTx Norm. DMSA J:43% b:57% Gr.s.40.kissúly pp: 2,3-3,3cm 6.hó:4,0-4,7cm 19é, GFR:93 j: 10,4cm b.: 7cm Gr.s.33.oligohydr. s.ces., Scr:313 Gr.s.37. retard. 6.hó oed.pulm.,VE 4,0-4,2cm (n<4,5) 27hó: veseTx (apa) Gr.s.34. +3ó pp Potter IIB Gr.s.40. +1n pp Potter IIB
 27hó: veseTx (apa) Gr.s ó pp. Potter IIB. Gr.s n pp. Potter IIB.")
18
Vese hypoplasiák, dysplasiák
Változatos megjelenés egy családon belül életképtelen magzat →tünetmentes gyermek tünetmentes felnőtt kisebb vesével →veseTx GN-t utánzó proteinuria Prenatalis nem felismerhető, de oligohydramnion Nincs vesecysta, VUR, extrarenalis tünet AD öröklés, de nincs mutatáció Paired box2 - PAX2 renal-coloboma Eyes absent1 -EYA1 brachio-oto-renal Hep.nucl.f.1b-HNF1b vesecysta és diabetes

19
FSGS klasszifikációja, okai
Primér: idiopath, permeabilitást indukáló cytokin? Secunder FSGS herediter podocyta defectus -actinin4, podocin, nephrin, CD2AP podocyta károsítás vírus: HIV, parvovírus B19, SV40 toxin: heroin, pamidronat postadaptív (hyperfilt.) visc.epithel nem osztódik hypertrophia nephronvesztés miatt reflux-analg. NP, foc.prol. GN, a.ren.sten., dysplasia, graft elégt., subtot.nephrect. hemodin. stress (vasodil.) norm. nephronszámon diabetes, sarlósejt, morbid obesitas,preeclampsia
 visc.epithel nem osztódik. hypertrophia nephronvesztés miatt reflux-analg. NP, foc.prol. GN, a.ren.sten., dysplasia, graft elégt., subtot.nephrect. hemodin. stress (vasodil.) norm. nephronszámon diabetes, sarlósejt, morbid obesitas,preeclampsia.")
20
Ismert fontosabb podocyta gének
protein gén funkció kórkép nephrin NPHS1 19q13 diaphragma felépítés jeltovábbítás CNF AR, FSGS-cong. podocin NPHS2 1q25 membran protein, jeltovábbítás SR-INS FSGS-AR CD2-assoc. protein CD2AP actinhoz köti a nephrint és podocint ? (FSGS-idiop.) α-actinin-4 ACTN4 19q13 cytoskeleton felépítése FSGS-AD felnőtt Lmx1B protein LMX1B 9q34,1 transzkripciós faktor köröm-patella WT1 fehérje WT1 11p13 IDM, Frasier, Denys-Drash
 α-actinin-4. ACTN4 19q13. cytoskeleton felépítése. FSGS-AD felnőtt. Lmx1B protein. LMX1B 9q34,1. transzkripciós faktor. köröm-patella. WT1 fehérje. WT1 11p13. IDM, Frasier, Denys-Drash.")
21
Miért kell a genetikai diagnózis?
Mutáció megtalálása magyarázatot ad a családnak Segít a következő generáció szűrésében A betegnek legtöbbször nem segít, de sokszor elkerülhető a felesleges/ártalmas vizsgálat, kezelés

22
Esetismertetés P.T. 30é nő 2000.febr: első terh. 37.h praeeclampsia, sectio ceas. PU: 4,4g/n, alb: 20, cr:69 2000.máj: PU:1,8g/n, vesebiopsia: FSGS jul.: 3x1g MP iv, majd 32mg po. aug.: PU: 4,2g/n, 3x50mg Cytoxan+16mg MP nov.: PU: 2,8g/n, 2x100mg CsA+ 4 mg MP 2001.márc.: PU: 3g/n, gyógyszerek ex! 2004.márc: subfebr., PU:12g/n, alb:25, chol:15, cr:80 3x1g MP iv, majd 64mg/n máj.: PU:5g/n, Imuran 3x50mg, MP:32mg jun.: PU: 5g/n, CsA 2x100mg majd 3x100mg szept.: PU:1,6g, cr: 114, gyógyszereket elhagyja 2004.okt: internetes chat, ben jelentkezik

23
Esetismertetés: az apa: P.E. 57é férfi
1962: sokizületi gyulladás, majd tonsillectomia 1966: szűrővizsgálat során proteinuria 1969: PU:+, ül:cyl.,1-2 vvt, residum tonsillect. 1971: PU:3+, vesebiopsia: membranas NP 150mg Imuran, heti 4x40mg prednisolon 1,5 év 1972: PU:0,9g/n, rebiopsia, indomethacin 1évig 1975: PU: 4+, Ccr: 100ml/p, RR: norm. vél.: állapota stacioner, th-t nem igényel gondozást abbahagyta, vizelet-vérvizsgálat nem volt szemészeti kezelés glaucoma miatt évek óta 2004.okt: PU: 6g/n, chol: 6,9, s-cr: 89

25
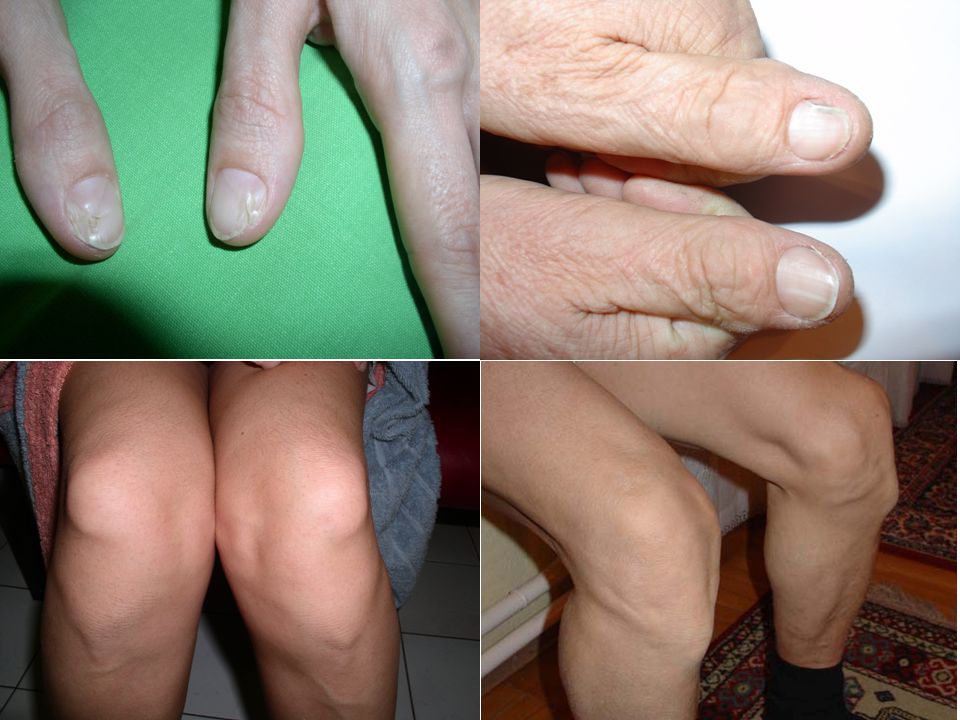
Köröm-patella syndroma
gyakoriság: 1: ? → kb. 200 beteg Mo-n orvosi irodalomban eddig több >500 beteg Little: Lancet 1897 (első közlés) Renwick JH: Ann Hum Genet 1955 (ABO vércsop) Sweeney E: J Med Gen 2003;40: (legnagyobb) 123 beteg, 43 család phenotípus jellemzése Trinn Cs és mtsai: Orv-Hetil 1996;137: AD, nagy penetráció, extrém változatos megjelenés a családok között és családon belül többség születéskor felismerhető, mégis családok generációi nem kerülnek felismerésre bár számos orvos, különböző szakorvos látja
 Renwick JH: Ann Hum Genet 1955 (ABO vércsop) Sweeney E: J Med Gen 2003;40: (legnagyobb) 123 beteg, 43 család phenotípus jellemzése. Trinn Cs és mtsai: Orv-Hetil 1996;137: AD, nagy penetráció, extrém változatos megjelenés. a családok között és családon belül. többség születéskor felismerhető, mégis. családok generációi nem kerülnek felismerésre. bár számos orvos, különböző szakorvos látja.")
26
ezekben a területekben csíkolt fibrilláris anyag
Vese hisztológia FM, IF: jellegtelen EM: focalis, változó GBM megvastagodás, többszörös, szabálytalan elektron-lucens területek; „molyrágta lyukak” ezekben a területekben csíkolt fibrilláris anyag hasonló elváltozás a mesangiumban is lehet Heidet L et al: Am J Patol 2003;163:

27
Esetismertetés: PT fia sz. 2000.
2004.okt. vizeletvizsgálat ált. és ül: negatív alb:126 (n<30), IgG:33 (n<8,5), b2m: <0,2 mg/l elfo: enyhe nem szelektív glomerularis proteinuria
, IgG:33 (n<8,5), b2m: <0,2 mg/l. elfo: enyhe nem szelektív glomerularis proteinuria.")
Hasonló előadás