Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
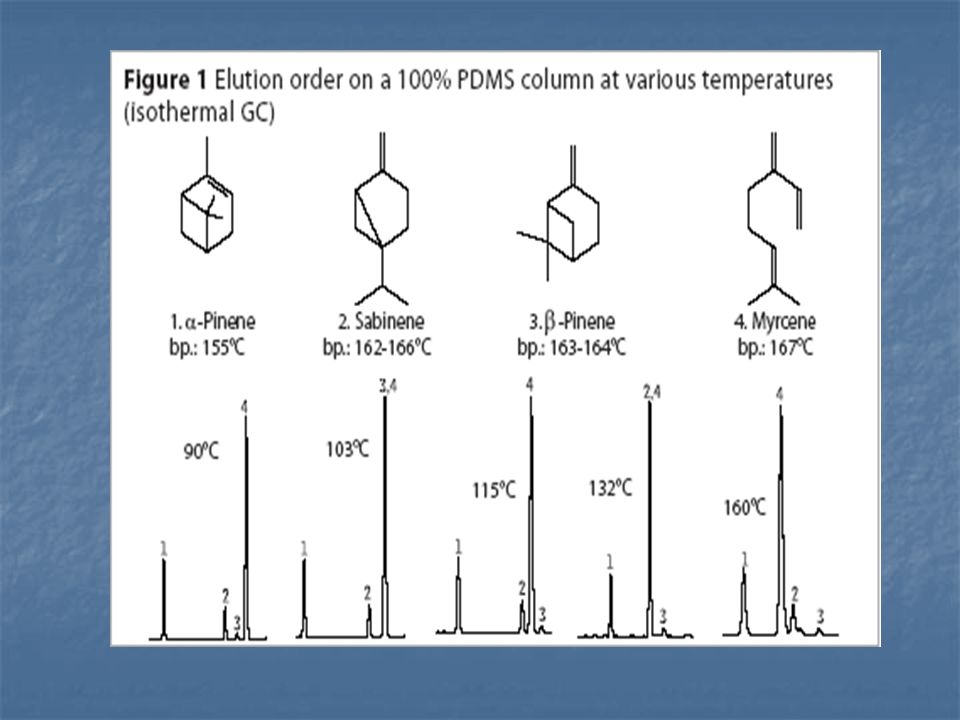
GÁZKROMATOGRÁFIA állófázis mozgófázis mechanizmus szilárd gáz
1952 James és Martin -gáz-folyadék kromatográfia; -Nobel díj a megoszlási kromatográfia kidolgozásáért. típus állófázis mozgófázis mechanizmus gáz-szilárd GSC szilárd gáz adszorpció gáz-folyadék GLC folyadék megoszlás

2
A gázkromatográfiás készülék
oszlop detektor gázpalack rekorder mintabemérő egység A gázkromatográfia bomlás nélkül gázzá alakítható, illékony vegyületek elválasztására alkalmas

3
Vívőgázrendszer Biztosítja a minta komponenseinek áthaladását az oszlopon Részei: Gázpalack (nyomáscsökkentő reduktor), N2, Ar (FID), H2, He Gáztisztító (aktív szenes, molekulaszűrős) Áramlásszabályozó - állandó áramlási sebesség kell, cm3/perc hőmérséklet nő → gáz viszkozítása nő → nyomásprogramozás oszlop töltet „öregszik” → ellenállása nő
 Áramlásszabályozó - állandó áramlási sebesség kell, cm3/perc. hőmérséklet nő → gáz viszkozítása nő → nyomásprogramozás. oszlop töltet „öregszik → ellenállása nő.")
4
Vívőgázok H (mm) Lineáris áramlási sebesség (cm/s)
A HETPmin értéke a H2 és a He esetén viszonylag széles lineáris gázsebesség tartományban csak kis mértékben változik, míg a N2 esetén szűk az a sebesség tartomány, ahol a HETP értéke a minimumhoz közeli. A gázkromatográfiás gyakorlatban a csúcsszélesedés csökkentése érdekében hőmérsékletprogramozást célszerű alkalmazni. A gázok, így a mozgófázisok belső súrlódása a hőmérséklet emelkedés hatására a moláris tömeggel arányosan nő, ami a lineáris gázsebesség állandó csökkenését eredményezi a kromatogram felvétele során. A fentiekből következően N2 alkalmazása csak izoterm méréseknél célszerű, míg a He és a H2 akár izoterm, akár hőmérsékletprogramozás esetén is használható. Töltetes oszlopok esetén biztonsági okokból a H2 alkalmazása nem ajánlott. Az alábbiak szerint a kapilláris oszlopoknál a legelőnyösebb tulajdonságokkal a H2 alkalmazható mozgófázisként: A legkisebb moláris tömeg és a legnagyobb diffúzió-sebesség miatt a legkisebb nyomás mellett állítható be az optimális lineáris sebesség. Nem teljesen inert gáz, reduktív hatása gátolja a káros oxidációs folyamatokat. Az analízisidő a legkisebb, mivel uopt értéke a H2-nél a legnagyobb. Lineáris áramlási sebesség (cm/s)
")
5
Mintaadagoló Feladata: gázból, vagy folyadékból gyorsan és dugószerűen, kis mennyiségeket ( μl gázoknál, 1-10 μl folyadékoknál) az oszlopra injektálni. Folyadékminta esetén biztosítani kell a gyors és teljes elgőzölögtetést → fűthető mintakamra ( C > oszlop T, 350 0C véghőmérséklet). Egyöntetű és bomlás nélküli elpárologtatás.
 az oszlopra injektálni. Folyadékminta esetén biztosítani kell a gyors és teljes elgőzölögtetést → fűthető mintakamra ( C > oszlop T, 350 0C véghőmérséklet). Egyöntetű és bomlás nélküli elpárologtatás.")
6
Gáz minták a, gázfecskendő b, gázbemérő hurok
minta be minta be minta ki minta ki oszlop vivőgáz vivőgáz

7
Injektálási technikák
1, Direkt az oszlopra – Flash injection Töltetes és wide bore (makrokapilláris) oszlopoknál Teljes minta mennyisége pillanatszerűen az oszlopra kerül.
 oszlopoknál. Teljes minta mennyisége pillanatszerűen az oszlopra kerül.")
8
2, Split injektálás – lefuvatás, megosztás
Kapilláris oszlopoknak kisebb a kapacitása. Split: Minta elgőzölögteté-se, vivőgázba adagolása, majd elágaztatása (pl. 1/100-1/1000 arányban → 0,01 μl és 0,001 μl injektálásának felel meg. Liner

9
Split arány Septum purge

10
3, Split-splitless (Grob) injektálás
Cél: nagyobb mintamennyiségek injektálása a kapil-láris oszlopra, a hatékonyság csökkenésének mini-malizálásával. Feltételek: Oldószer forráspontja legyen a legkisebb az elválasztandó anyagok között. Injektor T-je > a legnagyobb fp-ú anyag fp-jánál. Oszlop T-je az oldószer fp-jánál fokkal – kondenzáció az oszlop elején, állófázis telítődése. Ezek teljesülésével csak egy kis része az oldószernek kerül be az oszlopra és így koncentrálódik a minta!

11
Kivitelezés: Split ág 0,5 - 1 percig zárva (= splitless idő, vagy purge time),utána indul a lefuvatás, miközben az oszloptermosztát felfűtési programja is elindul és fp-juknak megfelelően eluálódnak a komponensek.

12
Split technika diszkriminál – split arány szerint
Problémák Split technika diszkriminál – split arány szerint Must be high enough to ensure a narrow sample band. If the split flow is too low (eg. 10mL/ min) the peaks will become very broad because it takes too long for the sample to exit the liner. Must be low enough to maintain sensitivity. If the split flow is too high, most of the sample will be vented to the atmosphere instead of entering the column. This can be useful when injecting concentrated samples, but small peaks in dilute samples could disappear altogether. Typical split flows are between 25 and 200 ml/ min. 20-200
 the peaks will become very broad because it takes too long for the sample to exit the liner. Must be low enough to maintain sensitivity. If the split flow is too high, most of the sample will be vented to the atmosphere instead of entering the column. This can be useful when injecting concentrated samples, but small peaks in dilute samples could disappear altogether. Typical split flows are between 25 and 200 ml/ min")
13
Forráspont diszkrimináció Splitless technika diszkriminál – splitless idő ( purge time) alapján
The top chromatogram is an example of what happens when the purge time is too short. As can be seen, C22 has not had enough time to vaporize completely. The sample has spent a little over 10sec in the injection port and has not gained enough energy from the liner to completely vaporize the hydrocarbons. There is much high boiling point discrimination.

14
4, Programozható hőmérsékletű injektor (Programable Temperature Vaporization- PTV)
Olyan split/splitless injektor, melyben a hőmér-séklet is programozható. Alacsony T-ről gyors felfűtés, mely hatására a komponensek fp-juknak megfelelő sorrendben párolognak el. Nagy mennyiségű minta injektálása lassan ( μl ). Oldószer lefuvatása az injektálás elején. Hő hatására bomló anyagok elválasztására is jó. Programable Temperature Vaporization
. Oldószer lefuvatása az injektálás elején. Hő hatására bomló anyagok elválasztására is jó. Programable Temperature Vaporization.")
15
Kriogén hűtés Liq. N2 -1500C Liq. CO2 – 500C Peltier hűtő hő-
cserélő: etil-alkohol+víz – szoba T-200C

16
Injektálás utáni fókuszálás
Kriofókuszálás – splitless idő alatti csúcskiszé-lesedés megakadályozására. 1, Az oszlop elején kondenzált oldószer telíti az állófázist és oldja a mintakomponenseket, ilyenkor elválasztás nem történik. 2, Splitless idő letelte után már nem érkezik to-vábbi oldószer az oszlopra. 3, Az oszlop felfűtés elindulásával elsőnek az oldó-szer távozik, keskeny csúcsba koncentrálva a mintát. 4, Retention gap – állófázis nélküli „előtét” oszlop

17
Anyaga: üvegcső, vagy saválló acél.
Kolonna Fajtái: 2-6 mm belső d-s, töltetes. 0,1-0,5 mm belső d-s kapilláris. 1, Töltetes kolonna (1-5 m) Anyaga: üvegcső, vagy saválló acél. Hordozója: diatomaföld (nagy mechanikai szilárdság, nagy fajlagos felület, kémiailag inert anyag) Megosztófolyadék + alacsony forráspontú oldószer + szilárd hordozó (0,1-0,3 mm-es átlagos szemcseméret) → oldószer elpárologtatása Kapacitásuk nagy (pár μl injektálható).
 Anyaga: üvegcső, vagy saválló acél. Hordozója: diatomaföld (nagy mechanikai szilárdság, nagy fajlagos felület, kémiailag inert anyag) Megosztófolyadék + alacsony forráspontú oldószer + szilárd hordozó (0,1-0,3 mm-es átlagos szemcseméret) → oldószer elpárologtatása. Kapacitásuk nagy (pár μl injektálható).")
18
2, Kapilláris kolonna Hossza: 10-100 m, átmérője:
1, <0,15 mm mikrokapilláris 2, 0,15-0,50 mm standard kapilláris 3, > 0,50 makrokapilláris (wide bore) Hordozó nincs. Anyaga kvarc. Poliamid bevonattal. A megosztófolyadékot közvetlenül a cső belső falára viszik fel (d = 0,1-10 μm), nyomás alatt préselik át. Élettartama növelhető, ha a megosztófolyadékot valamilyen kémiai kötéssel (szilanizálással) rögzítik. Kapacitásuk kicsi (μl törtrésze). Kapilláris = nyitottcső
 Hordozó nincs. Anyaga kvarc. Poliamid bevonattal. A megosztófolyadékot közvetlenül a cső belső falára viszik fel (d = 0,1-10 μm), nyomás alatt préselik át. Élettartama növelhető, ha a megosztófolyadékot valamilyen kémiai kötéssel (szilanizálással) rögzítik. Kapacitásuk kicsi (μl törtrésze). Kapilláris = nyitottcső.")
19
Megosztó (nedvesítő) folyadék = állófázis
Olyan makromolekuláris anyag, amely az elemzési T-n folyékony, de kellően hőstabil és gőztenziója is elhanyagolható. T max C < oszlop „vérzés” (bleeding) Hasonlóság elve: Apoláris komponensekhez apoláris Poláris komponensekhez poláris
 Hasonlóság elve: Apoláris komponensekhez apoláris. Poláris komponensekhez poláris.")
20
PLOT WCOT

21
Előtét oszlopok (guard column, retention gap)
Megosztó fázis nélküli kapilláris darab, mely adszorbeálja a minta szennyezőit. Retenciós idő függetlenítése az oszlop hosszától, szervízelés alatt. Kapilláris vágási éle

22
Gáz-szilárd kromatográfia (GSC) állófázisai
Szilikagél Aluminium-oxid Aktív szén Sztirol-divinil-benzol kopolimer Gáz-folyadék kromatográfia (GLC) Polisziloxán vázuak (szilikonok) Apoláris és poláris csoportokkal szubsztituált változatai mind apoláris, mind poláris vegyületek elválasztására alkalmasak. Szubsztituensek: alkil-, aril-, nitril-, vinil csoportok, vagy ezek keveréke.
 Polisziloxán vázuak (szilikonok) Apoláris és poláris csoportokkal. szubsztituált változatai mind apoláris, mind poláris vegyületek elválasztására alkalmasak. Szubsztituensek: alkil-, aril-, nitril-, vinil csoportok, vagy ezek keveréke.")
23
Előbbiek észterezett változatai, észterek elválasztására.
Fenilezett polisziloxán 2, Polietilén-glikolok Carbowax. Poláris álló-fázis. Polaritásuk a lánchossz növekedésével csökken. 3, Poliglikol-észterek Előbbiek észterezett változatai, észterek elválasztására. 2, polialkohol éterek

24
Kapilláris töltési technikák
dinamikus statikus In practice, a 5% w/w of stationary phase in the solvent will produce a film thickness of about 0.5 mm. However, this is only approximate, as the film thickness is also determined by the physical properties of the surface, the solvent and the stationary phase. The coating procedure is depicted in figure 15. After the plug has been run into the front of the column (sufficient to fill about 10% of the column length), pressure is applied to the front of the column to force the plug through the column at 2-4 mm per second (it will take about 5.5 hours for the plug to pass through a 60 m column). When the plug has passed through the column, the gas flow is continued for about an hour. The gas flow must not be increased too soon, or the stationary phase solution on the walls of the tube is displaced forward in the form of ripples, which produces a very uneven film. After an hour the flow rate can be increased and the column stripped of solvent. The last traces of the solvent are removed by heating the column above the boiling point of the solvent at an increased gas flow rate. Complete solvent removal can be identified by connecting the column to a detector and observing the baseline drift of the detector. After filling, one end of the column is sealed, and the other end is connected to a high vacuum pump and placed in an oven and the solvent slowly evaporates and the front retreats leaving a film of solution on the walls. The solvent then evaporates from this film and the stationary phase remains as a thin coating on the wall. The procedure is continued until all the solvent has evaporated and, except for the stationary phase, the column is empty. This process may take hours to complete. The procedure needs no attention and thus, can be carried out overnight. This procedure is more repeatable than the dynamic method of coating but, produces columns having similar performance to those dynamically coated.
, pressure is applied to the front of the column to force the plug through the column at 2-4 mm per second (it will take about 5.5 hours for the plug to pass through a 60 m column). When the plug has passed through the column, the gas flow is continued for about an hour. The gas flow must not be increased too soon, or the stationary phase solution on the walls of the tube is displaced forward in the form of ripples, which produces a very uneven film. After an hour the flow rate can be increased and the column stripped of solvent. The last traces of the solvent are removed by heating the column above the boiling point of the solvent at an increased gas flow rate. Complete solvent removal can be identified by connecting the column to a detector and observing the baseline drift of the detector. After filling, one end of the column is sealed, and the other end is connected to a high vacuum pump and placed in an oven and the solvent slowly evaporates and the front retreats leaving a film of solution on the walls. The solvent then evaporates from this film and the stationary phase remains as a thin coating on the wall. The procedure is continued until all the solvent has evaporated and, except for the stationary phase, the column is empty. This process may take hours to complete. The procedure needs no attention and thus, can be carried out overnight. This procedure is more repeatable than the dynamic method of coating but, produces columns having similar performance to those dynamically coated.")
25
Termosztát Az elválasztás hatékonyságát a megfelelő hőmérséklet program biztosítja. Célunk a minél jobb elválasztás (Rf > 1,5), minél gyorsabban. Komponensek növekvő forráspontjuk szerint eluálódnak. Fűtési tartomány: C Fűtési sebesség: 0,5-40 0C /perc Légtermosztát és gyors hűtés.
, minél gyorsabban. Komponensek növekvő forráspontjuk szerint eluálódnak. Fűtési tartomány: C. Fűtési sebesség: 0,5-40 0C /perc. Légtermosztát és gyors hűtés.")
26
Hőmérséklet programozott elúció

27
Hőmérséklet programozás
a, kezdeti T b, T ugrás c, végső T T nő → gázok oldhatósága csökken

28
T nő → gázok viszkozítása nő → u csökken
Ennek kompenzálására nyomásprogramozást kell alkalmazni a felfűtés során. Ez növeli a vivőgáz lineáris áramlási sebességét, és gyorsabb retenciós időket eredményez.

29
Módosított van Deemter egyenlet
Eredeti: H = 2 λdp + 2γDm k df2 u u π2 (1+k)2 Ds Módosított: H = B + Cm u + Cs u u Dm = a komponens diffúziós állandója a mozgófázis-ban u = az eluens lineáris áramlási sebessége df = a megosztófolyadék rétegvastagsága Ds = a komponens diffúziós állandója az állófázisban Cm ~ rc2/Dm
2 Ds. Módosított: H = B + Cm u + Cs u. u. Dm = a komponens diffúziós állandója a mozgófázis-ban. u = az eluens lineáris áramlási sebessége. df = a megosztófolyadék rétegvastagsága. Ds = a komponens diffúziós állandója az állófázisban. Cm ~ rc2/Dm.")
30
T hatása az elválasztásra

33
Oszlophossz hatása az elválasztásra

34
Az oszlopátmérő hatása az elválasztásra
Kapacitás csökken

35
A filmvastagság hatása
df a Van deemter egyenlet számlálojában van

36
Analízis idejének csökkentése a gyors elválasztás érdekében
Illékony minta Vékony állófázis (minőség!) Kis oszlop átmérő Rövidebb oszlop Hidrogén vívőgáz, nagy áramlási sebesség Meredek hőmérséklet gradiens
 Kis oszlop átmérő. Rövidebb oszlop. Hidrogén vívőgáz, nagy áramlási sebesség. Meredek hőmérséklet gradiens.")
37
Detektorok Feladatuk a kolonnából kilépő vívőgáz-áramban megjelenő komponensek folya-matos, gyors és érzékeny észlelése, az anyagmennyiséggel, vagy a koncentráció-val arányos jel szolgáltatása. Többféle, vagy egyszerre több detektor is alkalmazható! Szabályozott fűtés 400C-ig.

38
Detektorok jellemzése 1, Linearitási tartomány
Válaszjel Dinamikus tartomány Lineáris tartomány Mennyiség vagy koncentráció

39
2, Érzékenység Kalibrációs görbe meredeksége = d (válaszjel)
d (koncentráció) Közhasználatban a mérhető legkisebb koncentrációt is jelentheti! Kimutatási határ (limit of detection = LOD) A kimutatási határ a mért alkotónak az a legkisebb mennyisége, amely az adott módszerrel megbízhatóan megkülönböztethető a vak mintától. Megállapodás szerint, egy adott komponens: J LOD = J vak + 3 s vak J = válaszjel S vak = vakminta válaszjelének tapasztalati szórása Érzékenység Annak ellenére, hogy az érzékenység definíció szerint a detektor jel intenzitás– mintamennyiség függvény meredeksége, a gyakorlatban érzékenység alatt többnyire a detektorra és az adott komponensre jellemző MDL (minimum detectable limit = legkisebb detektálható mennyiség) értéket értjük. Általánosan elfogadott MDL érték a jel-zaj viszonyra 3:1 arány. Ez azt jelenti, hogy a legkisebb detektálható mennyiség által kapott csúcsmagasság a detektor elektronikus alapzaj értékének legalább háromszorosa. Egy adott detektor érzékenysége csak adott komponensre értelmezhető.
 Közhasználatban a mérhető legkisebb koncentrációt is jelentheti! Kimutatási határ (limit of detection = LOD) A kimutatási határ a mért alkotónak az a legkisebb mennyisége, amely az adott módszerrel megbízhatóan megkülönböztethető a vak mintától. Megállapodás szerint, egy adott komponens: J LOD = J vak + 3 s vak. J = válaszjel. S vak = vakminta válaszjelének tapasztalati szórása. Érzékenység. Annak ellenére, hogy az érzékenység definíció szerint a detektor jel intenzitás– mintamennyiség függvény meredeksége, a gyakorlatban érzékenység alatt többnyire a detektorra és az adott komponensre jellemző MDL (minimum detectable limit = legkisebb detektálható mennyiség) értéket értjük. Általánosan elfogadott MDL érték a jel-zaj viszonyra 3:1 arány. Ez azt jelenti, hogy a legkisebb detektálható mennyiség által kapott csúcsmagasság a detektor elektronikus alapzaj értékének legalább háromszorosa. Egy adott detektor érzékenysége csak adott komponensre értelmezhető.")
40
Kimutatási határ- LOD > 3×zaj Gyakorlatban
alapvonal zaj Kimutatási határ- LOD > 3×zaj Gyakorlatban 3:1 arány. Ez azt jelenti, hogy a legkisebb detektálható mennyiség által kapott csúcsmagasság a detektor elektronikus alapzaj értékének legalább háromszorosa. Egy adott detektor érzékenysége csak adott komponensre értelmezhető. alapvonal emelkedés

41
3, Szelektivítás Szelektivitás ill. specifikusság alatt értjük az adott detektor kiemelkedő érzékenységét a vegyületek bizonyos csoportjára. Specifikus detektorok azok, amelyek elemekre, elemek bizonyos csoportjára, funkciós csoportokra, vagy egyéb tulajdonságokra szelektíven adnak értékelhető jelet. Mátrix hatás csökkentése! Univerzális detektorok azok, amelyek minden, az oszlopról eluálódó komponensre értékelhető jelet szolgáltatnak. Ilyen a tömegspektrométer (MS Mass Spectormeter).
.")
42
Lángionizációs detektor
FLAME IONIZATION DETECTOR, (FID) Kollektor elektród The FID detector employs hydrogen as the combustion gas which is mixed with the column eluent (helium, nitrogen or other appropriate gas) and burnt at a small jet situated inside a cylindrical electrode. A potential of a few hundred volts is applied between the jet and the electrode and when a carbon containing solute is burnt in the jet, the electron/ion pairs that are formed are collected at the jet and cylindrical electrode. Az oszlopról eluálódó komponensek a lángionizációs detektorban (Flame ionization detector, FID) egy kb K hőmérsékletű hidrogén-levegő lángba jutnak. A lángban a C-H kötéseket tartalmazó molekulák, azaz a szerves vegyületek fragmentálódnak és egy részük ionizálódik. A képződött ionok a jet és a kollektor közötti potenciálkülönbség hatására a kollektora jutva a detektorba eluálódó anyagmennyiséggel arányos ionáramot eredményeznek ( Mikroégő - jet
 Kollektor elektród. The FID detector employs hydrogen as the combustion gas which is mixed with the column eluent (helium, nitrogen or other appropriate gas) and burnt at a small jet situated inside a cylindrical electrode. A potential of a few hundred volts is applied between the jet and the electrode and when a carbon containing solute is burnt in the jet, the electron/ion pairs that are formed are collected at the jet and cylindrical electrode. Az oszlopról eluálódó komponensek a lángionizációs detektorban (Flame ionization detector, FID) egy kb K hőmérsékletű hidrogén-levegő lángba jutnak. A lángban a C-H kötéseket tartalmazó molekulák, azaz a szerves vegyületek fragmentálódnak és egy részük ionizálódik. A képződött ionok a jet és a kollektor közötti potenciálkülönbség hatására a kollektora jutva a detektorba eluálódó anyagmennyiséggel arányos ionáramot eredményeznek ( Mikroégő - jet.")
43
Pirolízis: CnHm → n CH. + (m-n) H. Oxidáció: n CH. + n O. → n CHO.
A lángionizációs detektor egy kisméretű H2/levegő gáz-eleggyel táplált láng, amely fölé elektródpárt kapcsolnak. Az égés során a lángba bejutó szerves anyag először termikusan bomlik (pirolízis), utána oxidálódik, majd ionizálódik, mely lépésben a molekulák C atomszámával arányos számú e- keletkezik. Pirolízis: CnHm → n CH. + (m-n) H. Oxidáció: n CH. + n O. → n CHO. Ionizáció: n CHO. → n CHO+ + n e- The ionization process is not very efficient, only % of the solute molecules produce ions, (about two ions or electrons per 105 molecules). Nevertheless, because the noise level is very small, the minimum detectable mass of n-heptane is only 2 x g/sec. At a column flow rate of 20 ml/min. this is equivalent to a minimum detectable concentration of about 3 x g/ml.
, utána oxidálódik, majd ionizálódik, mely lépésben a molekulák C atomszámával arányos számú e- keletkezik. Pirolízis: CnHm → n CH. + (m-n) H. Oxidáció: n CH. + n O. → n CHO. Ionizáció: n CHO. → n CHO+ + n e- The ionization process is not very efficient, only % of the solute molecules produce ions, (about two ions or electrons per 105 molecules). Nevertheless, because the noise level is very small, the minimum detectable mass of n-heptane is only 2 x g/sec. At a column flow rate of 20 ml/min. this is equivalent to a minimum detectable concentration of about 3 x g/ml.")
44
Az ionok és e- képződése → gyenge áram, mely erősítés után mérhető és a komponens koncentrációjával (C atom-számával) arányos jelet szolgáltat. Érzékeny – g/anyag Széles linearitási tartomány: 107

45
Ideális esetben - hidrogén:(vivőgáz+make-up gáz) = 1:1
Kapilláris oszlopok esetén make-up gázt kell keverni a hidrogénhez (vivőgáz), hogy nagyobb jelet kapjunk, ez általában N2. Ideális esetben - hidrogén:(vivőgáz+make-up gáz) = 1:1 Nagyon széleskörben alkalmazott detektor. Majdnem univerzális, kivételek: Formaldehid, hangyasav, N2, O2, nemesgá-zok, CO, CO2, SO2, SO3, H2S, NO, NO2, NH3, HX és H2O. Reducing the dead volume. With earlier detectors the capillary column could not be installed close enough to the flame tip. With modern GC's it is installed 1 cm below the flame tip. If the column is too far removed, it has a major effect on the dead volume present. The column flow of a capillary column is too small to overcome this effect. The sample components take a long time to reach the flame causing the peaks to broaden (tailing). Increasing the sensitivity. The response of a FID is not only dependent on the flows of hydrogen and air, but also on the flow of carrier gas. A correct carrier gas flow rate can stabilize the flame, which leads to a reduction in noise level. The highest sensitivity can be obtained with approx. 30 mL/min of nitrogen. As the column cannot provide this flow, it is recommended to add make-up gas. Nitrogen is generally preferred as the make-up gas as it gives the highest sensitivity and is cheapest. Increasing the lifetime of the detector. Some detectors are more sensitive to overheating (and thus additional wear and tear) than other detectors. Especially when the gas flows are not optimized (wrong instrument adjustment, fused silica parts in the detector jet). Make-up gas provides enough cooling to prolong the detector lifetime.
, hogy nagyobb jelet kapjunk, ez általában N2. Ideális esetben - hidrogén:(vivőgáz+make-up gáz) = 1:1. Nagyon széleskörben alkalmazott detektor. Majdnem univerzális, kivételek: Formaldehid, hangyasav, N2, O2, nemesgá-zok, CO, CO2, SO2, SO3, H2S, NO, NO2, NH3, HX és H2O. Reducing the dead volume. With earlier detectors the capillary column could not be installed close enough to the flame tip. With modern GC s it is installed 1 cm below the flame tip. If the column is too far removed, it has a major effect on the dead volume present. The column flow of a capillary column is too small to overcome this effect. The sample components take a long time to reach the flame causing the peaks to broaden (tailing). Increasing the sensitivity. The response of a FID is not only dependent on the flows of hydrogen and air, but also on the flow of carrier gas. A correct carrier gas flow rate can stabilize the flame, which leads to a reduction in noise level. The highest sensitivity can be obtained with approx. 30 mL/min of nitrogen. As the column cannot provide this flow, it is recommended to add make-up gas. Nitrogen is generally preferred as the make-up gas as it gives the highest sensitivity and is cheapest. Increasing the lifetime of the detector. Some detectors are more sensitive to overheating (and thus additional wear and tear) than other detectors. Especially when the gas flows are not optimized (wrong instrument adjustment, fused silica parts in the detector jet). Make-up gas provides enough cooling to prolong the detector lifetime.")
46
Silica deposition results in a white deposit formed throughout the entire detector. It is the result of frequemt analysis of silylated samples. Careful cleaning of the detector according to the manufacturer's instructions will readily solve this problem. Soot deposition, the 'black problem', is the result of the analysis of large series of samples containing solvents that do not readily burn, such as halogenated solvents, CS2, toluene etc. Also when cleaning, in particular of the flame tip, will solve this problem. Oxidation of the collector electrode, the 'green problem', is caused by oxidative species such as HCL or HF formed when chlorinated or fluorinated solvents are used. This problem will require replacement of the detector. Incorrect flow settings can result in similar problems as contamination. Since checking the gas flows is very simple, this should always be done before attempting to clean the detector.

47
Elektronbefogási detektor ELECTRON CAPTURE DETECTOR (ECD)
béta-sugárzás tipikusan a neutron felesleggel rendelkező atommagok bomlási módja. Ekkor ugyanis egy neutron átalakul protonná, miközben egy elektron keletkezik. Az így felszabaduló energia jelentős részét az elektron mozgási energiája viszi el. Az atomból nagy sebességgel kilépő elektron a béta-részecske. A béta-bomlás során tehát az atom rendszáma egyel nő, tömegszáma viszont változatlan marad

48
Specifikus ionszelektiv detektor, mely a halogéntartalmú (F, Cl, Br) növényvédő- és rovarirtószer maradványok meghatározásában nélkülözhetetlen. Negatív pólus: 63 Ni természetes β-sugárzó izotópot tartalmazó fólia az elektródba építve = sugárforrás. Pozítiv pólus: kollektor elektród, melynek irányába haladnak az e- -ok. Az így létrejött polarizációs feszültség csak néhány volt (1-10V) → A állandó alapionáramot hoz létre. A 63Ni‑fóliából emittálódó ‑részecskék energiája 67 keV, ezek a ‑részecskék ütköznek a vivőgáz atomjaival. A rugalmas és rugalmatlan ütközések révén gyökökből, pozitív ionokból (,, Ar+,…) és termikus elektronokból álló plazma jön létre, mely nem rendelkezik akkora energiával, hogy a nagy ionizációs potenciálú szervetlen és szerves molekulákat ütközés révén ionizálja. Ezek az elektronok az anód (kollektor; pozitív pólus) felé haladva zárják az áramkört. Az áramvezetésben a pozitív ionok nem vesznek részt mivel nagy a tömegük, így kicsi a mozgékonyságuk az elektronokhoz képest.
 → A állandó alapionáramot hoz létre. A 63Ni‑fóliából emittálódó ‑részecskék energiája 67 keV, ezek a ‑részecskék ütköznek a vivőgáz atomjaival. A rugalmas és rugalmatlan ütközések révén gyökökből, pozitív ionokból (,, Ar+,…) és termikus elektronokból álló plazma jön létre, mely nem rendelkezik akkora energiával, hogy a nagy ionizációs potenciálú szervetlen és szerves molekulákat ütközés révén ionizálja. Ezek az elektronok az anód (kollektor; pozitív pólus) felé haladva zárják az áramkört. Az áramvezetésben a pozitív ionok nem vesznek részt mivel nagy a tömegük, így kicsi a mozgékonyságuk az elektronokhoz képest.")
49
Ha nagy elektronegativitású elemeket (F, Cl, Br, ,O) tartalmazó molekulák jutnak a detektorba → abszorbeálják az e- -okat → alapionáram csökken, mivel nagy tömegű és kis mozgékonyságú anionok képződnek. Leggyakrabban nitrogén vivőgázzal használják.

50
Halogénmentes oldószerek használata kötelező!
Lehetővé teszi a halogéntartalmú gázkromatográ-fiásan vizsgálható vegyületek mérését 10‑12‑10‑15 g‑ig. A detektor szénhidrogénekre gyakorlatilag érzéketlen, nagy elektronvonzó‑képességű csopor-tot (pl. -NO2, konjugált rendszerek) tartalmazó vegyületek specifikus mérésére alkalmas. Linearitási tartománya 3‑4 nagyságrend fluor és klór tartalmú vegyületekre. Make-up gáz használata szükséges, hogy gyorsabban kiürüljön a detektor tér (N2,Ar+CH4). Halogénmentes oldószerek használata kötelező! A detektor alkalmazásánál ügyelni kell arra, hogy csak halogénmentes oldószereket használjunk, mert a nagy érzékenység miatt még igen kis szennyezés is zavarja a mérést. A GC‑ECD rendszerrel történő méréshez alkalmazott halogénmentes oldószereket lehetőleg olyan helyiségben kell tárolni, ahol nem fordulnak elő klórozott oldószerek. Actually, the mechanism of operation of the ECD is slightly more complex. The electrons emitted by the radioactive foil move too fast to be captured by the analyte molecules. They have to be slowed down. This can be achieved by introducing an inert gas into the ECD cell that collides with the rapid electrons and in that way slowing them down. For this reason some percentage of methane (5%) is added to the make-up gas argon (the most sensitive gas for an ECD regarding its ionizing capability). Methane reduces the electron energy by collision without ionizing the methane and avoids interference effects such as component ionization. Today, nitrogen (cheaper and universal) is often used as make-up gas for an ECD instead of the more specific argon/methane.
 tartalmazó vegyületek specifikus mérésére alkalmas. Linearitási tartománya 3‑4 nagyságrend fluor és klór tartalmú vegyületekre. Make-up gáz használata szükséges, hogy gyorsabban kiürüljön a detektor tér (N2,Ar+CH4). Halogénmentes oldószerek használata kötelező! A detektor alkalmazásánál ügyelni kell arra, hogy csak halogénmentes oldószereket használjunk, mert a nagy érzékenység miatt még igen kis szennyezés is zavarja a mérést. A GC‑ECD rendszerrel történő méréshez alkalmazott halogénmentes oldószereket lehetőleg olyan helyiségben kell tárolni, ahol nem fordulnak elő klórozott oldószerek. Actually, the mechanism of operation of the ECD is slightly more complex. The electrons emitted by the radioactive foil move too fast to be captured by the analyte molecules. They have to be slowed down. This can be achieved by introducing an inert gas into the ECD cell that collides with the rapid electrons and in that way slowing them down. For this reason some percentage of methane (5%) is added to the make-up gas argon (the most sensitive gas for an ECD regarding its ionizing capability). Methane reduces the electron energy by collision without ionizing the methane and avoids interference effects such as component ionization. Today, nitrogen (cheaper and universal) is often used as make-up gas for an ECD instead of the more specific argon/methane.")
51
Organochloride insecticides
8. Endocrine disruptor, xenoestrogen endosulfan DDT was the first modern pesticide and is arguably the best known organic pesticide. It is a highly hydrophobic colorless solid with a weak, chemical odor that is nearly insoluble in water but has a good solubility in most organic solvents, fat, and oils. DDT is also known under the chemical names 1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane and dichloro-diphenyl-trichloroethane (from which the abbreviation was derived). DDT was developed as the first of the modern insecticides early in World War II. It was initially used with great effect to combat mosquitoes spreading malaria, typhus, and other insect-borne human diseases among both military and civilian populations, and as an agricultural insecticide. The Swiss chemist Paul Hermann Müller of Geigy Pharmaceutical in Switzerland was awarded the Nobel Prize in Physiology or Medicine in 1948 "for his discovery of the high efficiency of DDT as a contact poison against several arthropods." In 1962, American biologist Rachel Carson published the book Silent Spring, which alleged that DDT caused cancer and harmed bird reproduction by thinning egg shells.[1] The book resulted in a large public outcry which eventually led to the insecticide being banned for agricultural use in the USA, and was one of the signature events in the birth of the environmental movement. DDT was subsequently banned for agricultural use in many countries in the 1970s; there is still a great controversy regarding the impact of this decision on the use of DDT to fight disease vectors. DDT
ethane and dichloro-diphenyl-trichloroethane (from which the abbreviation was derived). DDT was developed as the first of the modern insecticides early in World War II. It was initially used with great effect to combat mosquitoes spreading malaria, typhus, and other insect-borne human diseases among both military and civilian populations, and as an agricultural insecticide. The Swiss chemist Paul Hermann Müller of Geigy Pharmaceutical in Switzerland was awarded the Nobel Prize in Physiology or Medicine in 1948 for his discovery of the high efficiency of DDT as a contact poison against several arthropods. In 1962, American biologist Rachel Carson published the book Silent Spring, which alleged that DDT caused cancer and harmed bird reproduction by thinning egg shells.[1] The book resulted in a large public outcry which eventually led to the insecticide being banned for agricultural use in the USA, and was one of the signature events in the birth of the environmental movement. DDT was subsequently banned for agricultural use in many countries in the 1970s; there is still a great controversy regarding the impact of this decision on the use of DDT to fight disease vectors. DDT.")
52
Két dimenziós GC (GC×GC)
")
53
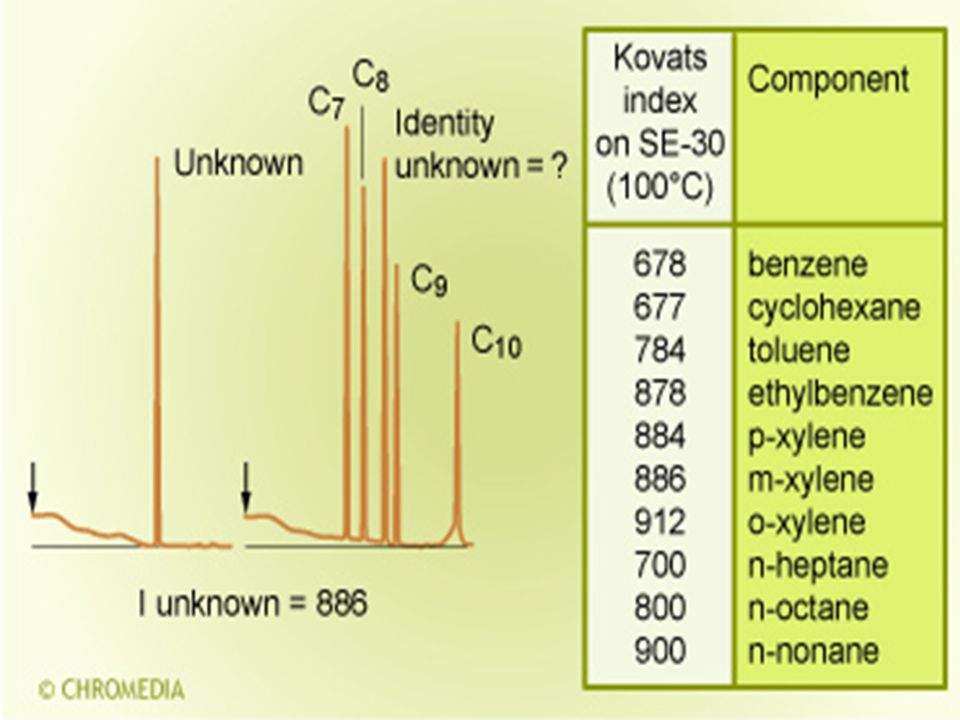
Minőségi azonosítás Minőségi azonosításra a retenciós időt, illetve a relatív retenciós időt használjuk. Kováts-féle retenciós index – szerves vegyületek homológ sorain belül az egyes alkotók redukált retenciós idejének a logaritmusa a C atomszám lineáris függvénye. I = 100n.

54
n-alkánok homológ sora
lg t’R C’ lg t’R n+1 C lg t’Rx A lg t’Rn B B’ 700 Ix 800 I

55
A Kováts féle retenciós index adott vegyületre, adott állófázison, izoterm körülmények között független az oszlop méreteitől, a nyomáseséstől és az áramlási sebességtől. Apoláris állófázison mért retenciós index egyenes arányban van a forrásponttal, tehát a vegyület forráspontja közelítőleg meghatározható. Poláris alkotók indexének hőmérsékletfüggése nem mindig lineáris. Hasonló szerkezetű vegyületeknél azonos helyettesítések a retenciós indexet azonos mértékben változtatják meg.

57
Kalibrációs módszer - külső standard módszer
Mennyiségi elemzés Kalibrációs módszer - külső standard módszer Különböző ismert koncentrációjú oldat kromatográfiás csúcs alatti területét mérjük, majd ezek felhasználá-sával kalibrációs egyenest készítünk, melynek iránytangense az érzékenység. Ismeretlen minta koncentrációja az általa szolgáltatott terület / az érzékenység: Kalibrációs egyenes képlete Ai: az i-edik komponens csúcsterülete, wi az i-edik komponens mennyisége

58
Deca-BDE = 10 Br atom egy biszfenil-éteren.

59
Belső standard módszer
Egy ún. belső standardot - mindig azonos mennyiségben hozzáadunk, mind a kalibráló oldatokhoz, mind a mintákhoz. A relatív érzékenység (fi) használatán alapszik. Előnye, hogy kiküszöböli a térfogatos mintabevitelből adódó hibát (a minta komponenseinek teljes és pillanatszerű elpárologtatásának hiánya). Kalibrációs egyenes képlete Ai = ai + bi wi AS As a belső-standard csúcsterülete az aktuális kromatogramban, wi az i-edik komponens mennyisége a belső standard mennyiségével osztva.
 használatán alapszik. Előnye, hogy kiküszöböli a térfogatos mintabevitelből adódó hibát (a minta komponenseinek teljes és pillanatszerű elpárologtatásának hiánya). Kalibrációs egyenes képlete. Ai. = ai + bi wi. AS. As a belső-standard csúcsterülete az aktuális kromatogramban, wi az i-edik. komponens mennyisége a belső standard mennyiségével osztva.")
61
Alkalmazhatóságának feltételei
A mintában biztosan ne forduljon elő. Kémia és fizikai tulajdonságai hasonlóak legyenek a meghatározandó komponensekéihez. A minta egyik komponensével se lépjen fel együttes elúció (koelúció).
.")
62
Származékképzési reakciók
Célja: a mérendő komponens átalakítása valamilyen speciális kémiai reakcióval, annak érdekében, hogy a keletkezett termék könnyen gázkromatografálható legyen. Illékonyság és termikus stabilitás növelése. H-hídképzés csökkentése → csúcsalak javítása Átalakítandó funkciós csoportok: - OH, - COOH, -NH2, - SH, stb. LOD értékek csökkentése – ECD detektornál : halogéntartalmú származékképzés

63
Származékképzés hatása

64
Legfőbb típusaik Szililezés Alkilezés Acilezés 1, SZILILEZÉS
Aktív hidrogén lecserélése általában trimetil-szilil csoportra. Általánosan: 3 Reakció hatásfoka a szililezőszer erősségétől és a kémiai reakcióban résztvevő funkciós csoport minőségétől függ.

65
Szililezhetőségi sorrend:
Alkoholok > Fenolok > Karbonsavak > Aminok > Amidok > Tiolok Sztérikus hatások: Primer alkoholok > szekunder alkoholok > tercier alkoholok Reakció feltételei: A reagenst feleslegben alkalmazzuk. 100 %-osnak kell lennie az átalakulásnak. Reakció körülmények optimálása ( C, 10 – 120 perc, katalizátor). Lehetőleg egyfajta származék keletkezzen (több funkciós csoport esetén), több csúcs keletkezése probléma!
. Lehetőleg egyfajta származék keletkezzen (több funkciós csoport esetén), több csúcs keletkezése probléma!")
66
Leggyakrabban alkalmazott szililezőszerek reaktivítási sorrendben
N-trimetilszilil-imidazol TMSI N,O-bisz-trimetilszilil-trifluoracetamid BSTFA N,O-bisz-trimetilszilil-acetamid BSA N-metil-N-trimetilszilil-trifluoracetamid MSTFA Hexametil-diszilazán HMDS TMSI aminokkal nem reagál- aminosavaknál szelektív származékolás, acetamid: CH3CONH2 BSTFA illékonyabb, minta a BSA és kevésbé tömíti el a FID detektort MSTFA a legillékonyabb illékony vegyületek elemzéséhez jó

67
Xvégső = NH3 Reakciómechanizmus: Nukleofil szubsztitució Kivitelezés: Oldószer nélkül, csak a szililezőszerrel Oldószerrel (aprotikus): piridin, toluol, acetonitril, hexán Katalizátorok: trifluoro-ecetsav (TFA), trimetil-klór-szilán (TMCS) Egyéb követelmények: Szililezőszerek nagytisztaságúak és vízmentesek legyenek és ne zavarják az elválasztást (koelució mentes). Származékolandó minta vízmentessége! Könnyen hidrolizálnak víz és alkoholok hatására. Stabilitás vizsgálat.
: piridin, toluol, acetonitril, hexán. Katalizátorok: trifluoro-ecetsav (TFA), trimetil-klór-szilán (TMCS) Egyéb követelmények: Szililezőszerek nagytisztaságúak és vízmentesek legyenek és ne zavarják az elválasztást (koelució mentes). Származékolandó minta vízmentessége! Könnyen hidrolizálnak víz és alkoholok hatására. Stabilitás vizsgálat.")
68
Aktív hidrogéneket alkil csoportra cseréljük.
2, ALKILEZÉS Aktív hidrogéneket alkil csoportra cseréljük. Pl. Alkohol → Éter; Karbonsav → Észter Fajtái: 1, Alkilhalogenidek (jodidok, bromidok) R-OH + CH3-I → R-O-CH3 + HI Fenolok, karbonsavak, tiolok Katalizátor: ezüst-oxid 2, Diazotálás (diazometánnal, mérgező) R-OH + CH2N2 → R-O-CH3 + N2 Fenolok, karbonsavak, szulfonsavak Diazometán felesleg elpárologtatása
 R-OH + CH3-I → R-O-CH3 + HI. Fenolok, karbonsavak, tiolok. Katalizátor: ezüst-oxid. 2, Diazotálás (diazometánnal, mérgező) R-OH + CH2N2 → R-O-CH3 + N2. Fenolok, karbonsavak, szulfonsavak. Diazometán felesleg elpárologtatása.")
69
3, ACILEZÉS Aktív hidrogéneket acil csoportra cseréljük Pl. Alkoholok → Észter, Aminok → Savamid Savanhidridek és savkloridok használata Legáltalánosabban használt reagensek: 1, trifluoroecetsav-anhidrid (TFAA) 2, pentafluoropropionsav-anhidrid (PFPA) 3, heptafluorobutánsav-anhidrid (HFBA)
 2, pentafluoropropionsav-anhidrid (PFPA) 3, heptafluorobutánsav-anhidrid (HFBA)")
70
A halogén atomok bevitelével lehetőség nyílik az ECD detektor használatára, ami érzékenység növekedést jelent. A reakciót vízmentes körülmények között hajtják végre, majd ezt követi a reagens feleslegének eltávolítása bepárlással, vagy gyenge bázis vizes oldatával való elreagáltatással. A fluorozott származékoknak nagyobb az illékonysága, mint a többi perhalogénezett származéké.

71
A GÁZKROMATOGRÁFIA ANALITIKAI ALKALMAZÁSA
Kőolajipar, petrolkémiai ipar Környezetvédelmi analízis Gyógyszeripari elemzések Oldószermaradványok elemzése Élelmiszer analitika (aromaanyagok, toxinok, szermaradványok) Kozmetikai ipar (illatanyagok).
 Kozmetikai ipar (illatanyagok).")
72
Régészet - A manchesteri múzeum munkatársai egy múmia pólyáját vizsgálták kromatográfiás eljárással, és méhviaszt, bitument, növényi eredetű galbánummézgát és tamarinduszkivonatot mutattak ki. A vizsgálatok szerint a külső rétegeket állati eredetű ragasztóval rögzítették, noha a feljegyzések szerint a mumifikáláshoz nem használtak állati eredetű anyagokat.

73
Egy mocsaras, neolitikumi településről, a mai Németország területéről származó cseréptöre-déken, csont- és kőszerszámon fekete vagy barna, amorf, kátrányszerű lerakódást fedeztek fel. A vizsgálatok kimutatták, hogy számos lelet pentaciklusos triterpént tartalmaz, és minden oldószeres extraktum legfontosabb komponense a betulin volt. Ismert, hogy ezek a triterpének a nyírfa (Betula pendula) külső kérgében is előfordulnak. Az ősi kátrány tehát nyírfaké-regből származik. Ragasztónak és rágóguminak használták.
 külső kérgében is előfordulnak. Az ősi kátrány tehát nyírfaké-regből származik. Ragasztónak és rágóguminak használták..")
75
Albedo grapefruit szerves komponenseinek trimetilszilil(oxim)-származékainak GC-MS felvétele, hidrolízis nélkül (A) és hidrolízissel (B) 1. Foszforsav 2. Levulinsav 3. Almasav 4. Aszparaginsav 5. Glutaminsav 6. Trimetoxibenzoesav 7. Alanin 8. Prolin 9. 4-hidroxi-benzoesav 10. Xilóz 20. Galakturonsav 21. Inozitol 22. Szedoheptulóz 23. Cukor-foszfátok 24., 25., 27.,28. diszacharidok 26. Szacharóz 29. Naringenin 30. Hesperetin 31. Klorogénsav 32. Tokoferol 33. Rozmaringsav A * 11. Arabinóz 12. Vanilinsav 13. Ramnóz 14. Fukóz 15. Citromsav 16. Kínasav 17. Fruktóz 18. Glükóz 19. Galaktóz B 18. This compilation shows the elution profile of the constituents of grapefruit albedos before and after hydrolysis, determined as their trimethyl (oxime) ether/ester derivatives, by gas chromatography-mass spectrometry: partly on the basis of the total ions of derivatives, partly on the basis of their selective fragment ions * Zs. Füzfai, I. Molnár-Perl, J. Chromatogr. A, 2007, in press Pittcon-2007
 ether/ester derivatives, by gas chromatography-mass spectrometry: partly on the basis of the total ions of derivatives, partly on the basis of their selective fragment ions. * Zs. Füzfai, I. Molnár-Perl, J. Chromatogr. A, 2007, in press. Pittcon")
76
Melamin és társai Melamine is a nitrogen-rich industrial chemical that became well-known in North America in 2007 after its presence in wheat gluten was linked with renal failure in dogs and cats. A large pet food recall occurred in 2007 when animals became ill after eating contaminated food, some animals eventually died. There was a suspected link between the sick animals and melamine contamination, although melamine was believed to have low or no toxicity.2 Melamine is an industrial chemical used for the production of plastics, adhesives, flame retardants, fabrics and other materials.3 Melamine is not a food ingredient but was found to be present in pet food and other protein-containing goods. It is believed that the toxicity is due to the combination of melamine and cyanuric acid forming insoluble crystals. The crystals can form in the kidneys, causing sickness and eventual renal failure.4 As the investigation continued, it was determined that melamine and melamine byproducts were intentionally added This is done to artificially elevate the protein content values of products. The Kjeldahl method is used to determine protein content. This method works by testing for nitrogen content. When protein is digested, nitrogen is released and converted to ammonia. The amount of ammonia is determined by titration and is correlated to the amount of protein present. This method yields falsely high numbers when nonprotein nitrogen is in the sample. Melamine and melamine production byproducts were added to protein products to gain higher protein content. The structures of melamine and related compounds, pictured below, show very high nitrogen content, which make them ideal as nonprotein nitrogen sources. Unfortunately, the melamine production byproducts contain not only melamine, but also cyanuric acid, ammeline, and ammelide, which cause toxicity in pets. Due to the adverse effects of melamine contamination, imported products need to be tested for the presence of melamine, cyanuric acid and related compounds.

77
Macskatáp elemzése (50µg/g spike)
Column:Rtx®-5MS, 30m, 0.25mm ID, 0.25µm Sample:melamine (4), cyanuric acid (1), ammelide, ammeline, benzoguanamine (IS) (10µg/mL prederivatized) Inj.:1µL, splitless (hold 1 min.), 3.5mm splitless inlet liner Inj. temp.:280°C Carrier gas:helium,Flow rate:1mL/min. Oven temp.:75°C to 320°C 15°C/min. (hold 4 min.)
, cyanuric acid (1), ammelide, ammeline, benzoguanamine (IS) (10µg/mL prederivatized) Inj.:1µL, splitless (hold 1 min.), 3.5mm splitless inlet liner Inj. temp.:280°C. Carrier gas:helium,Flow rate:1mL/min. Oven temp.:75°C to 320°C 15°C/min. (hold 4 min.)")
Hasonló előadás




 1/26 Energia és környezet NO x keletkezés és kibocsátás.>")












>")
>")
>")
