Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
Tömegspektrometria A tömegspektrometria olyan vizsgálati módszer, amelynél ionos részecskéket választunk el fajlagos tömegük (töltésegységre eső tömegük: m/z) szerint, csökkentett nyomáson, elektromos, vagy mágneses mezők segítségével. Az elválasztott ionok intenzitását folyamatosan mérjük, s így egy ionáram intenzitás - fajlagos tömeg függvény-kapcsolathoz, az ún. tömeg-spektrumhoz jutunk. Ez a tömegspektrum a minőségi információ alapja, ugyanis nincs két olyan szerves vegyület, amelyiknek a tömegspektruma azonos lenne.
 szerint, csökkentett nyomáson, elektromos, vagy mágneses mezők segítségével. Az elválasztott ionok intenzitását folyamatosan mérjük, s így egy ionáram intenzitás - fajlagos tömeg függvény-kapcsolathoz, az ún. tömeg-spektrumhoz jutunk. Ez a tömegspektrum a minőségi információ alapja, ugyanis nincs két olyan szerves vegyület, amelyiknek a tömegspektruma azonos lenne.")
2
Tömegspektrométer részei
Mintabeviteli rendszer (közvetlen: gáz, folyadék, vagy szilárd minta bevitele, közvetett: GC, HPLC. Ionforrás az ionoptikával (ionok előállítása). Analizátor (ionok elválasztása fajlagos tömegük szerint). Detektor (ion, vagy fotonsokszorozó). Vákuumrendszer (első fokozat egy olajrotációs szivattyú (0,1 kPa), a második egy turbomole-kuláris pumpa, mellyel kPa nyomást lehet elérni). Számítógép szabályzó és adatkezelő (adatgyűjtő, feldolgozó, értékelő, archíváló) funkcióval. Az ionforrás feladata, hogy a vizsgálandó molekulából valamilyen gerjesztõ energia (kinetikus, fény, elektromos, kémiai, stb.) segítségével ionokat hozzon létre és ezeket az ionokat lehetõleg azonos kinetikus energiával, egy nyalábban mozgatva, gyorsítva juttassa az analizátorba. A koherens ionnyaláb létrehozását az ún. ionoptika biztosítja, s általában néhány kilovoltos feszültségkülönbség mozgatja a nyaláb ionjait. Az alkalmazott gerjesztési energiától függõen többféle ionforrás létezik. A leggyakoribb az elektronütközéses (EI: electron inpact) ionforrás, amely 50-75 eV energiájú termikus elektronokkal hoz létre ütközési ionizációt gáz fázisban. (Az adatbankokban összegyűjtött tömegspektrumok 95 %-a ilyen ionforrással készült.) Ez a gázfázisú ionizáció behatárolja a vizsgálható vegyületek körét is, hiszen ha bomlás nélkül nem párologtatható el az adott vegyület, akkor nem is vizsgálható e módszerrel. Más esetekben ugyan elpárologtatható a molekula bomlás nélkül, de nem stabil a molekulaionja, azaz keletkezésekor azonnal elbomlik, vagy nagyon kicsi az intenzitása. Kíméletesebb ionizációs megoldást jelent a kémiai-, a gyors atom bombázásos (FAB), a foto-, a lézer deszorpciós, az elektromos, stb. ionizáció. A kémiai ionizációs ionforrás összetett ionforrás. Egy EI ionforrásban reagns gáz (metán, ammónia, stb.) molekulákból ionokat állít elő, amelyeket gyorsítással juttat a hozzá közvetlenül kapcsolódó térrészbe, ahová a vizsgálandó minta gázfázisú molekuláit vezetik be. Az analizátor választja el az ionforrásból nagy sebességgel érkezõ ionokat fajlagos tömegük szerint. Az elválasztás többféle elv alapján oldható meg. Így megkülönböztethetünk: 1. repülési idő (TOF: time of flight), 2. elektromos, pl. kvadrupól, radiofrekvenciás, ioncsapda, omegatron, stb., 3. mágneses analizátorú, 4. elektrosztatikus, illetve 5. kettõs fókuszálású (nagy felbotóképességű), illetve 6. tandem analizátorokat (MS/MS, MSn) alkalmazó tömegspektrométereket. Lényeges eleme a tömegspektrométerek működésének a vákuumot biztosító, többnyire legalább kétfokozatú vákuumrendszer. Az első fokozatot többnyire olajrotációs szivattyú biztosítja, amely atmoszférikusról 3 nagyságrendnyi nyomáscsökkenést hoz létre. Ez biztosítja az olajdiffúziós, vagy turbomolekuláris szivattyúzás elővákuumát. A végvákuum kPa, amelyet a korszerű rendszerekkel néhány óra alatt el lehet érni és folyamatosan biztosítani lehet. Erre szükség is van, mivel az ionforrásban keletkezõ ionoktól várjuk, hogy a tömegspektrum jellegét megszabják. Ez csak akkor biztosítható, ha a primer, monomolekuláris folyamatokban keletkezõ ionok a gyorsítást követően egymással reakcióba nem léphetnek. Ehhez van szükség a nagy vákuumra, amelynek adott hőmérsékleten olyannak kell lennie, hogy a részecskék szabad úthossza nagyobb legyen, mint az az út, amit a keletkezésüktõl a detektálásukig (elhalásukig) kénytelenek megtenni.
. Analizátor (ionok elválasztása fajlagos tömegük szerint). Detektor (ion, vagy fotonsokszorozó). Vákuumrendszer (első fokozat egy olajrotációs szivattyú (0,1 kPa), a második egy turbomole-kuláris pumpa, mellyel kPa nyomást lehet elérni). Számítógép szabályzó és adatkezelő (adatgyűjtő, feldolgozó, értékelő, archíváló) funkcióval. Az ionforrás feladata, hogy a vizsgálandó molekulából valamilyen gerjesztõ energia (kinetikus, fény, elektromos, kémiai, stb.) segítségével ionokat hozzon létre és ezeket az ionokat lehetõleg azonos kinetikus energiával, egy nyalábban mozgatva, gyorsítva juttassa az analizátorba. A koherens ionnyaláb létrehozását az ún. ionoptika biztosítja, s általában néhány kilovoltos feszültségkülönbség mozgatja a nyaláb ionjait. Az alkalmazott gerjesztési energiától függõen többféle ionforrás létezik. A leggyakoribb az elektronütközéses (EI: electron inpact) ionforrás, amely eV energiájú termikus elektronokkal hoz létre ütközési ionizációt gáz fázisban. (Az adatbankokban összegyűjtött tömegspektrumok 95 %-a ilyen ionforrással készült.) Ez a gázfázisú ionizáció behatárolja a vizsgálható vegyületek körét is, hiszen ha bomlás nélkül nem párologtatható el az adott vegyület, akkor nem is vizsgálható e módszerrel. Más esetekben ugyan elpárologtatható a molekula bomlás nélkül, de nem stabil a molekulaionja, azaz keletkezésekor azonnal elbomlik, vagy nagyon kicsi az intenzitása. Kíméletesebb ionizációs megoldást jelent a kémiai-, a gyors atom bombázásos (FAB), a foto-, a lézer deszorpciós, az elektromos, stb. ionizáció. A kémiai ionizációs ionforrás összetett ionforrás. Egy EI ionforrásban reagns gáz (metán, ammónia, stb.) molekulákból ionokat állít elő, amelyeket gyorsítással juttat a hozzá közvetlenül kapcsolódó térrészbe, ahová a vizsgálandó minta gázfázisú molekuláit vezetik be. Az analizátor választja el az ionforrásból nagy sebességgel érkezõ ionokat fajlagos tömegük szerint. Az elválasztás többféle elv alapján oldható meg. Így megkülönböztethetünk: 1. repülési idő (TOF: time of flight), 2. elektromos, pl. kvadrupól, radiofrekvenciás, ioncsapda, omegatron, stb., 3. mágneses analizátorú, 4. elektrosztatikus, illetve. 5. kettõs fókuszálású (nagy felbotóképességű), illetve. 6. tandem analizátorokat (MS/MS, MSn) alkalmazó tömegspektrométereket. Lényeges eleme a tömegspektrométerek működésének a vákuumot biztosító, többnyire legalább kétfokozatú vákuumrendszer. Az első fokozatot többnyire olajrotációs szivattyú biztosítja, amely atmoszférikusról 3 nagyságrendnyi nyomáscsökkenést hoz létre. Ez biztosítja az olajdiffúziós, vagy turbomolekuláris szivattyúzás elővákuumát. A végvákuum kPa, amelyet a korszerű rendszerekkel néhány óra alatt el lehet érni és folyamatosan biztosítani lehet. Erre szükség is van, mivel az ionforrásban keletkezõ ionoktól várjuk, hogy a tömegspektrum jellegét megszabják. Ez csak akkor biztosítható, ha a primer, monomolekuláris folyamatokban keletkezõ ionok a gyorsítást követően egymással reakcióba nem léphetnek. Ehhez van szükség a nagy vákuumra, amelynek adott hőmérsékleten olyannak kell lennie, hogy a részecskék szabad úthossza nagyobb legyen, mint az az út, amit a keletkezésüktõl a detektálásukig (elhalásukig) kénytelenek megtenni.")
3
Ionforrások Feladata: a vizsgálandó molekulából valamilyen gerjesztő energia (kinetikus, fény, elektromos, kémiai, stb.) segítségével ionokat hozzon létre és ezeket az ionokat lehetőleg azonos kinetikus energiával, egy nyalábban mozgatva, gyorsítva juttassa az analizátorba. 1, Elektronütközési (elektronimpakt) ionizáció (EI) Leggyakoribb (95%) 50-75 eV energiájú termikus elektronok (wolfrámizzószál) Ütközési ionizáció gázfázisban Az ionforrás feladata, hogy a vizsgálandó molekulából valamilyen gerjesztõ energia (kinetikus, fény, elektromos, kémiai, stb.) segítségével ionokat hozzon létre és ezeket az ionokat lehetõleg azonos kinetikus energiával, egy nyalábban mozgatva, gyorsítva juttassa az analizátorba. A koherens ionnyaláb létrehozását az ún. ionoptika biztosítja, s általában néhány kilovoltos feszültségkülönbség mozgatja a nyaláb ionjait. Az alkalmazott gerjesztési energiától függõen többféle ionforrás létezik. A leggyakoribb az elektronütközéses (EI: electron inpact) ionforrás, amely 50-75 eV energiájú termikus elektronokkal hoz létre ütközési ionizációt gáz fázisban. (Az adatbankokban összegyűjtött tömegspektrumok 95 %-a ilyen ionforrással készült.) Ez a gázfázisú ionizáció behatárolja a vizsgálható vegyületek körét
 segítségével ionokat hozzon létre és ezeket az ionokat lehetőleg azonos kinetikus energiával, egy nyalábban mozgatva, gyorsítva juttassa az analizátorba. 1, Elektronütközési (elektronimpakt) ionizáció (EI) Leggyakoribb (95%) eV energiájú termikus elektronok (wolfrámizzószál) Ütközési ionizáció gázfázisban. Az ionforrás feladata, hogy a vizsgálandó molekulából valamilyen gerjesztõ energia (kinetikus, fény, elektromos, kémiai, stb.) segítségével ionokat hozzon létre és ezeket az ionokat lehetõleg azonos kinetikus energiával, egy nyalábban mozgatva, gyorsítva juttassa az analizátorba. A koherens ionnyaláb létrehozását az ún. ionoptika biztosítja, s általában néhány kilovoltos feszültségkülönbség mozgatja a nyaláb ionjait. Az alkalmazott gerjesztési energiától függõen többféle ionforrás létezik. A leggyakoribb az elektronütközéses (EI: electron inpact) ionforrás, amely eV energiájú termikus elektronokkal hoz létre ütközési ionizációt gáz fázisban. (Az adatbankokban összegyűjtött tömegspektrumok 95 %-a ilyen ionforrással készült.) Ez a gázfázisú ionizáció behatárolja a vizsgálható vegyületek körét.")
4
EI ionforrás V tér V = V 1: mintabevezető nyílás; 2: ionvisszaverő lemez (repeller); 3: izzószál; 4: elektronbevezető nyílás; 5 és 6: iongyorsító rés; 7: belépő nyílás; 8: ionképződés helye; 9: anód
; 3: izzószál; 4: elektronbevezető nyílás; 5 és 6: iongyorsító rés; 7: belépő nyílás; 8: ionképződés helye; 9: anód.")
5
Etilbenzol spektruma Relatív intenzitás bázis csúcs molekulaion

6
2, Kémiai ionizáció (CI) Kíméletesebb ionizáció Fragmensek számának csökkentése (tömegspektrum egyszerűsítése). A mérendő mintát az elektronforrásba belépése előtt "reagens" gázzal kb. tízezer-szeresére hígítják. A reagens gáz molekulái ionizálódnak. Az így keletkezett primer ionok ütközése a vizsgá-landó molekulákkal vezet a szekunder ionok képződéséhez.

7
Kémiai ionizáció főbb lépései metán gáz esetén
Primer ionképződés: Szekunder ionképződés: Pszeudo-molekulaion képződése

8
Szilárd hordozóra felvitt minták vizsgálata
1, Gyors atom ütközési ionizáció – Fast Atom Bombardment (FAB) Argonágyú (4-10 keV) → szilárd hordozóra felvitt minta, vagy szilárd minta → felületéről szekunder ionok kilökése → analizátor. 2, Folyadék szekunder ion tömegspektrometria - Liquid Secondary Ion Mass Spectrometry (LSIMS) Céziumion ágyú (2-30 keV) → lsd. FAB Hordozó: fém Mátrix: glicerin, 3-nitro-benzil-alkohol Hőérzékeny vegyületek kíméletes ionizációja Egy EI forrásban Ar, vagy He atomok ionizálásával és gyorsításával keletkeznek gyors Ar+, vagy He+ ionok, amelyek egy ütközési kamrában semleges Ar, vagy He atomokkal ütközve átadják kinetikus energiájukat a semleges atomoknak. Ezek közül mindig elindul elegendő számú gyors atom abban az irányban, ahol a vizsgálandó minta molekuláival közvetlenül, vagy valamilyen mátrix közvetítésével ütközve, a minta molekulái kíméletesen ionizálódnak. Többnyire ilyenkor is pszeudo molekulaionok keletkeznek. Így az ilyen FAB-spektrum is kevés ionból áll, de a molekulaion (pszeudo) “megfogható”.
 Argonágyú (4-10 keV) → szilárd hordozóra felvitt minta, vagy szilárd minta → felületéről szekunder ionok kilökése → analizátor. 2, Folyadék szekunder ion tömegspektrometria - Liquid Secondary Ion Mass Spectrometry (LSIMS) Céziumion ágyú (2-30 keV) → lsd. FAB. Hordozó: fém. Mátrix: glicerin, 3-nitro-benzil-alkohol. Hőérzékeny vegyületek kíméletes ionizációja. Egy EI forrásban Ar, vagy He atomok ionizálásával és gyorsításával keletkeznek gyors Ar+, vagy He+ ionok, amelyek egy ütközési kamrában semleges Ar, vagy He atomokkal ütközve átadják kinetikus energiájukat a semleges atomoknak. Ezek közül mindig elindul elegendő számú gyors atom abban az irányban, ahol a vizsgálandó minta molekuláival közvetlenül, vagy valamilyen mátrix közvetítésével ütközve, a minta molekulái kíméletesen ionizálódnak. Többnyire ilyenkor is pszeudo molekulaionok keletkeznek. Így az ilyen FAB-spektrum is kevés ionból áll, de a molekulaion (pszeudo) megfogható .")
9
FAB és LSIMS [M+H]+ [M+Na]+
The techniques of FAB and LSIMS are very similar in concept and design as they both involve the bombardment of a solid spot of the analyte/matrix mixture on the end of a sample probe by a fast particle beam (see fig.1). The matrix (a small organic species like glycerol or 3-nitro benzylalcohol) is used to keep a homogenous sample surface. The particle beam is incident onto the surface of the analyte/matrix spot, where it transfers its energy bringing about localised collisions and disruptions. Some species are ejected (sputtered) from the surface as secondary ions by this process. These ions are then extracted and focussed before passing to the mass analyser. The polarity of ions produced depends on the source potentials - the figure shows a positive ion beam being formed. In FAB, the particle beam is a neutral inert gas (Ar or Xe) at 4-10 keV and in LSIMS, the particle beam is ions (usually Cs+) at 2-30 keV. Both methods are comparatively 'soft' ionisation methods - very little residual energy is possessed by the ions after desorption - making them particularly suited to the analysis of low volatility analytes. The resulting spectra consist largely of intact molecular species (e.g. [M+H]+ and [M+Na]+) with some minor structural fragmentation. The low mass region of the spectra are, however, dominated by matrix and matrix/salt cluster ions.
![FAB és LSIMS [M+H]+ [M+Na]+](http://slideplayer.hu/slide/2215315/8/images/9/FAB+%C3%A9s+LSIMS+%5BM%2BH%5D%2B+%5BM%2BNa%5D%2B.jpg "The techniques of FAB and LSIMS are very similar in concept and design as they both involve the bombardment of a solid spot of the analyte/matrix mixture on the end of a sample probe by a fast particle beam (see fig.1). The matrix (a small organic species like glycerol or 3-nitro benzylalcohol) is used to keep a homogenous sample surface. The particle beam is incident onto the surface of the analyte/matrix spot, where it transfers its energy bringing about localised collisions and disruptions. Some species are ejected (sputtered) from the surface as secondary ions by this process. These ions are then extracted and focussed before passing to the mass analyser. The polarity of ions produced depends on the source potentials - the figure shows a positive ion beam being formed. In FAB, the particle beam is a neutral inert gas (Ar or Xe) at 4-10 keV and in LSIMS, the particle beam is ions (usually Cs+) at 2-30 keV. Both methods are comparatively soft ionisation methods - very little residual energy is possessed by the ions after desorption - making them particularly suited to the analysis of low volatility analytes. The resulting spectra consist largely of intact molecular species (e.g. [M+H]+ and [M+Na]+) with some minor structural fragmentation. The low mass region of the spectra are, however, dominated by matrix and matrix/salt cluster ions.")
10
Alkalmazás: peptidek és más biomolekulák, szulfonsav-származékok, ionos szerves fémvegyületek.
FAB az egyik legjobb ionizációs technika ionos vegyületekre. Etilén-glikol-monoéter

11
Mátrix közvetítésével végzett lézer deszorpciós ionizáció - Matrix Assisted Laser Desorption Ionization (MALDI) “Fotoionizációs” – jól szabályozható lézerforrás → mátrix molekulák (fotodisszociáció elkerülése) → vizsgált molekulák Kíméletes ionizáció Termikusan érzékeny anyagok, enzimek, hormonok, vagy akár több százezer dalton tömegű biomolekulák, fehérje szekvenciák tömegspektrometriás vizsgálata. MALDI is a form of laser desorption ionisation. The sample is mixed with a saturated solution of matrix (an organic compound with a strong absorption at the laser wavelength) and a drop deposited on the MALDI target. Following solvent evaporation and matrix crystallisation, the target is placed in the mass spectrometer source and irradiated with pulses of laser light. There is a transfer of energy between excited matrix molecules and sample molecules, and both desorb from the condensed state. Once in the vapour phase, proton transfer between matrix and sample takes place - resulting in ion formation. Ions are accelerated out of the source by application of a high potential (c. 20 kV) to a series of extraction electrodes and lenses. Molecular species commonly observed include: [M+H]+, [M+NH4]+ and [M+Na]+ (positive ion mode), and [M-H]- (negative ion mode). MALDI is a relatively soft ionisation technique, and little fragmentation is observed. Because of its tendency to produce singly-charged ions (even with large proteins), MALDI is normally used in conjunction with mass analysers possessing high m/z capability, such as time of flight (TOF).
 → vizsgált molekulák. Kíméletes ionizáció. Termikusan érzékeny anyagok, enzimek, hormonok, vagy akár több százezer dalton tömegű biomolekulák, fehérje szekvenciák tömegspektrometriás vizsgálata. MALDI is a form of laser desorption ionisation. The sample is mixed with a saturated solution of matrix (an organic compound with a strong absorption at the laser wavelength) and a drop deposited on the MALDI target. Following solvent evaporation and matrix crystallisation, the target is placed in the mass spectrometer source and irradiated with pulses of laser light. There is a transfer of energy between excited matrix molecules and sample molecules, and both desorb from the condensed state. Once in the vapour phase, proton transfer between matrix and sample takes place - resulting in ion formation. Ions are accelerated out of the source by application of a high potential (c. 20 kV) to a series of extraction electrodes and lenses. Molecular species commonly observed include: [M+H]+, [M+NH4]+ and [M+Na]+ (positive ion mode), and [M-H]- (negative ion mode). MALDI is a relatively soft ionisation technique, and little fragmentation is observed. Because of its tendency to produce singly-charged ions (even with large proteins), MALDI is normally used in conjunction with mass analysers possessing high m/z capability, such as time of flight (TOF).")
12
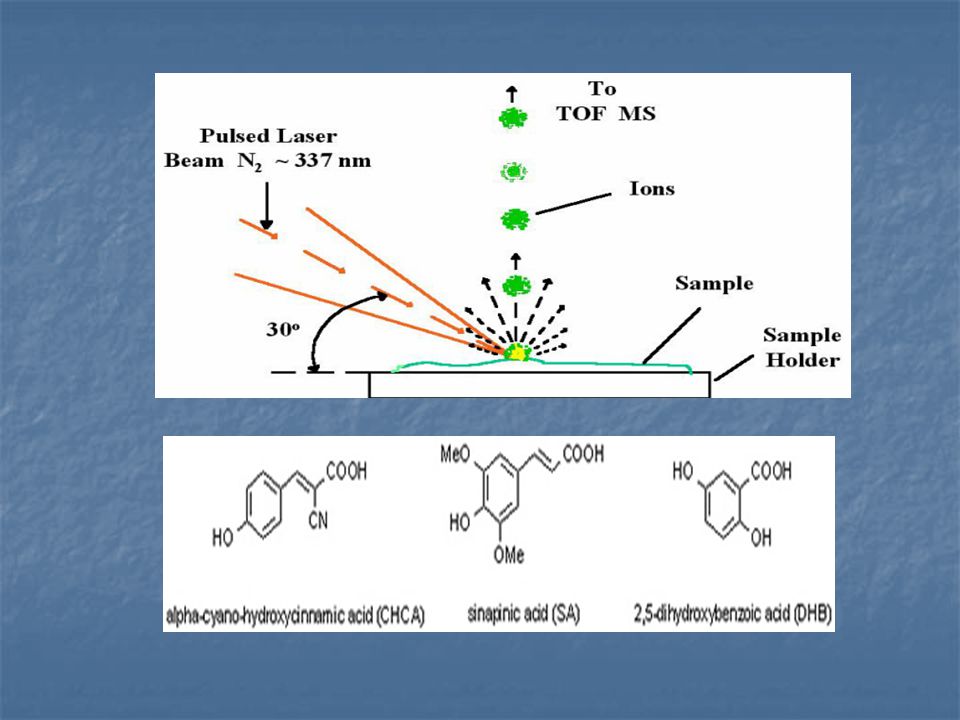
MALDI Most MALDI sources are equipped with a UV laser - excitation with the 337 nm band of a nitrogen laser being particularly common. Derivatives of benzoic and cinnamic acids are widely used as matrices, as they have both the required UV absorption properties and sufficient polarity to promote proton transfer between matrix and sample. For positive ion operation, 0.1 % TFA is sometimes added to the sample to promote ionisation. However, its use in negative ion MALDI should be avoided, as it suppresses sample ion formation. N2 lézer – 337 nm, UV Mátrix (benzoesav és fahéjsav származékok, 100x feleslegben) és minta beszárítása vákuumban Mátrix adszorbeálja a lézerfényt → elpárologtatja és ionizálja a mintamolekulákat
 és minta beszárítása vákuumban. Mátrix adszorbeálja a lézerfényt → elpárologtatja és ionizálja a mintamolekulákat.")
14
A Bovine Serum Albumin (BSA) MALDI spektruma
 MALDI spektruma")
15
Folyadékok ionizálási módszerei HPLC-interfész technikák
Termoszpré (Thermospray) – vákuumba porlasztás, hőstabil vegyületek. Elektroszpré (Electrospray) - atmoszférikus nyomáson folyadék porlasztás, hőérzékeny és ionizálható vegyületek. Atmoszférikus nyomású kémiai ionizáció Atmospheric Pressure Chemical Ionization (APCI) – atmoszférikus nyomáson folyadék porlasztás, hőstabil és nem ionos vegyületek, kémiai ionizáció. LÁGY IONIZÁCIÓ, KISMÉRTÉKŰ FRAGMENTÁCIÓ
 – vákuumba porlasztás, hőstabil vegyületek. Elektroszpré (Electrospray) - atmoszférikus nyomáson folyadék porlasztás, hőérzékeny és ionizálható vegyületek. Atmoszférikus nyomású kémiai ionizáció Atmospheric Pressure Chemical Ionization (APCI) – atmoszférikus nyomáson folyadék porlasztás, hőstabil és nem ionos vegyületek, kémiai ionizáció. LÁGY IONIZÁCIÓ, KISMÉRTÉKŰ FRAGMENTÁCIÓ.")
16
ELECTROSPRAY (ESI) Elektrospray ionizáció során a kromatografáló oszlopról eluálódó ionos molekulákat (szerves savak, szerves bázisok, vagy sóik) egy porlasztó gáz egy feszültség alatt lévő kapillárison porlaszt keresztül. A folyadékcseppekbe zárt töltéshordozók sűrűsége az intenzív párolgás (nincs hőbomlás!) követ-keztében egyre nagyobb lesz, s szinte szétrob-bannak a cseppek, s a szabaddá vált ionok gyorsítás után az analizátorban elválaszthatók fajlagos tömegük szerint.
 egy porlasztó gáz egy feszültség alatt lévő kapillárison porlaszt keresztül. A folyadékcseppekbe zárt töltéshordozók sűrűsége az intenzív párolgás (nincs hőbomlás!) követ-keztében egyre nagyobb lesz, s szinte szétrob-bannak a cseppek, s a szabaddá vált ionok gyorsítás után az analizátorban elválaszthatók fajlagos tömegük szerint.")
17
ELECTROSPRAY In electrospray ionisation (ESI) a fine spray of charged droplets is created by the application of a high voltage (typically 1-3 kV) to a capillary containing a flowing liquid. The process is often assisted by use of a co-axial nebuliser gas, such as nitrogen, but it is important to note that formation of the micro-droplets is ultimately an electrostatic rather than mechanical phenomenon (see below). As with APCI, electrospray ionisation occurs at atmospheric pressure, but solution processes - rather than gas phase ion/molecule reactions - result in ion formation 1-3 kV
 a fine spray of charged droplets is created by the application of a high voltage (typically 1-3 kV) to a capillary containing a flowing liquid. The process is often assisted by use of a co-axial nebuliser gas, such as nitrogen, but it is important to note that formation of the micro-droplets is ultimately an electrostatic rather than mechanical phenomenon (see below). As with APCI, electrospray ionisation occurs at atmospheric pressure, but solution processes - rather than gas phase ion/molecule reactions - result in ion formation. 1-3 kV.")
18
Pozitív ionizáció Negatív ionizáció Amino Karboxil Amid Hidroxil/fenol
In a simplified model of ESI, charged droplets - expelled from the tip of the capillary - evaporate until the Rayleigh limit is reached (i.e. the point at which Coulomb repulsion equals surface tension). Beyond this limit the droplets are unstable and explode to form micro-droplets. This process is repeated until individual solvated ions are formed. Evaporation of the solvent results in the generation of isolated gas-phase ions. Larger molecules, with a number of chargeable sites, tend to show a distribution of charge states. In the following illustration, the droplets contain singly, doubly and triply charged positive ions: ESI is suitable for the analysis of organic compounds with medium - high polarity. Since positive ionisation is dependent on protonation, molecules containing basic functional groups work well in this mode. Negative ionisation, in contrast, functions by deprotonation, thus the presence of acidic functional groups is a prerequisite for reasonable limits of detection. Although ESI has traditionally been used in the analysis of polar biomolecules, such as peptides, carbohydrates etc., many relatively small organics are amenable to this technique - providing they contain sufficient functionality. Pozitív ionizáció Negatív ionizáció Amino Karboxil Amid Hidroxil/fenol Észter Imid Aldehid/keto
. Beyond this limit the droplets are unstable and explode to form micro-droplets. This process is repeated until individual solvated ions are formed. Evaporation of the solvent results in the generation of isolated gas-phase ions. Larger molecules, with a number of chargeable sites, tend to show a distribution of charge states. In the following illustration, the droplets contain singly, doubly and triply charged positive ions: ESI is suitable for the analysis of organic compounds with medium - high polarity. Since positive ionisation is dependent on protonation, molecules containing basic functional groups work well in this mode. Negative ionisation, in contrast, functions by deprotonation, thus the presence of acidic functional groups is a prerequisite for reasonable limits of detection. Although ESI has traditionally been used in the analysis of polar biomolecules, such as peptides, carbohydrates etc., many relatively small organics are amenable to this technique - providing they contain sufficient functionality. Pozitív ionizáció. Negatív ionizáció. Amino. Karboxil. Amid. Hidroxil/fenol. Észter. Imid. Aldehid/keto.")
19
Pozitív ionizáció:[M+H]+, [M+Na]+, [M+CH3CN+H]+
Negatív ionizáció:[M-H]- [M+HCOO]- Lassú áramlási sebesség < 1 ml/min Eluens pH: - Savas, a bázikus komponenseknél, pozitív módban. - Bázisos, a savas komponenseknél, negatív módban. Puffer: Illékony és kis koncentrációjú (ion szupresszió elkerülése) The concentration of the buffer, or acid or base used to adjust/control the pH should be as low as possible. If not, competition between analyte and electrolyte ions for conversion to gas-phase ions decreases the analyte response.This can be explained as follows: if a species is in large excess, it will cover the droplet surface and prevent other ions to access the surface, and thus to evaporate. A species in large excess will also catch all charges available and prevent the ionisation of other molecules present at much lower concentration.
![Pozitív ionizáció:[M+H]+, [M+Na]+, [M+CH3CN+H]+](http://slideplayer.hu/slide/2215315/8/images/19/Pozit%C3%ADv+ioniz%C3%A1ci%C3%B3%3A%5BM%2BH%5D%2B%2C+%5BM%2BNa%5D%2B%2C+%5BM%2BCH3CN%2BH%5D%2B.jpg "Negatív ionizáció:[M-H]- [M+HCOO]- Lassú áramlási sebesség < 1 ml/min. Eluens pH: - Savas, a bázikus komponenseknél, pozitív módban. - Bázisos, a savas komponenseknél, negatív módban. Puffer: Illékony és kis koncentrációjú (ion szupresszió elkerülése) The concentration of the buffer, or acid or base used to adjust/control the pH should be as low as possible. If not, competition between analyte and electrolyte ions for conversion to gas-phase ions decreases the analyte response.This can be explained as follows: if a species is in large excess, it will cover the droplet surface and prevent other ions to access the surface, and thus to evaporate. A species in large excess will also catch all charges available and prevent the ionisation of other molecules present at much lower concentration.")
20
Lószív mioglobin ESI tömegspektruma (M: 16955 Da)
Gauss görbe Although protein digestion is still an important technique for mass spectrometry, it is now relatively easy to obtain direct mass measurements of proteins by ESI-MS. The multiple charges (see above) are statistically distributed upon the basic sites of the protein. Fig.3 shows a typical ESI mass spectrum of horse heart myoglobin (16.9 kDa). The peaks observed are due to the multiple charging affect, in this case the charges are roughly Gaussian distributed around the +15 charge state ranging from +22 to +10. The actual distribution of charges is dependent on a number of factors, but most commonly the electrospray conditions can greatly affect charge distribution as well as the gross structure of the protein (i.e. the actual availability of the basic sites). Studies of charge distribution are commonly used to make inferences about the tertiary structure of the protein.
 are statistically distributed upon the basic sites of the protein. Fig.3 shows a typical ESI mass spectrum of horse heart myoglobin (16.9 kDa). The peaks observed are due to the multiple charging affect, in this case the charges are roughly Gaussian distributed around the +15 charge state ranging from +22 to +10. The actual distribution of charges is dependent on a number of factors, but most commonly the electrospray conditions can greatly affect charge distribution as well as the gross structure of the protein (i.e. the actual availability of the basic sites). Studies of charge distribution are commonly used to make inferences about the tertiary structure of the protein.")
21
APCI Az APCI eredetileg nem ionos vegyületek vizsgá-latára alkalmas ionizációs interfész. A folyadékkromatográfiás eluens molekulái ionizálódnak (pl. a vízből keletkeznek H3O+), s ezek az ionok képesek protonálni (kémiai ioni-záció) az eredetileg nemionos molekulákat. A keletkező pszeudo-molekulaionokat (M+H)+, (M+Na)+, (M+oldószer ionja)+ és néhány fragmen-sét gyorsítás után az analizátor választja szét. Poláros szerves vegyületek analízise (gyógyszerek!) The electric field is sufficiently strong to ionise solvent vapour by either removal (positive ion mode) or donation (negative ion mode) of an electron. Ion/molecule reactions then result in the formation of a reactive species.
, s ezek az ionok képesek protonálni (kémiai ioni-záció) az eredetileg nemionos molekulákat. A keletkező pszeudo-molekulaionokat (M+H)+, (M+Na)+, (M+oldószer ionja)+ és néhány fragmen-sét gyorsítás után az analizátor választja szét. Poláros szerves vegyületek analízise. (gyógyszerek!) The electric field is sufficiently strong to ionise solvent vapour by either removal (positive ion mode) or donation (negative ion mode) of an electron. Ion/molecule reactions then result in the formation of a reactive species.")
22
APCI The significant difference is that APCI occurs at atmospheric pressure and has its primary applications in the areas of ionisation of low mass pharmaceutical compounds (APCI is not suitable for the analysis of thermally labile compounds). The general source set-up (see fig. 1) shares a strong resemblance to electrospray ionisation (ESI) and as such is most commonly used in conjunction with HPLC or other flow separation techniques. Where APCI differs to ESI, is in the way ionisation occurs. In ESI, ionisation is bought about through the potential difference between the spray needle and the cone along with rapid but gentle desolvation. In APCI, the analyte solution is introduced into a pneumatic nebulizer and desolvated in a heated quartz tube before interacting with the corona discharge creating ions. St. Elmo’s Fire = korona kisülés St. Elmo's Fire and normal sparks both can appear when high electrical voltage affects a gas. St. Elmo's fire is seen during thunderstorms when the ground below the storm is electrically charged, and there is high voltage in the air between the cloud and the ground. The voltage tears apart the air molecules and the gas begins to glow. It takes about 30,000 volts per centimeter of space to start a St. Elmo's fire (although sharp points can trigger it at somewhat lower voltage levels.) The nitrogen and oxygen in the earth's atmosphere causes St. Elmo's Fire to fluoresce with blue or violet light; this is similar to the mechanism that causes neon lights to glow[5]. N2 400 0C 3 kV
. The general source set-up (see fig. 1) shares a strong resemblance to electrospray ionisation (ESI) and as such is most commonly used in conjunction with HPLC or other flow separation techniques. Where APCI differs to ESI, is in the way ionisation occurs. In ESI, ionisation is bought about through the potential difference between the spray needle and the cone along with rapid but gentle desolvation. In APCI, the analyte solution is introduced into a pneumatic nebulizer and desolvated in a heated quartz tube before interacting with the corona discharge creating ions. St. Elmo’s Fire = korona kisülés St. Elmo s Fire and normal sparks both can appear when high electrical voltage affects a gas. St. Elmo s fire is seen during thunderstorms when the ground below the storm is electrically charged, and there is high voltage in the air between the cloud and the ground. The voltage tears apart the air molecules and the gas begins to glow. It takes about 30,000 volts per centimeter of space to start a St. Elmo s fire (although sharp points can trigger it at somewhat lower voltage levels.) The nitrogen and oxygen in the earth s atmosphere causes St. Elmo s Fire to fluoresce with blue or violet light; this is similar to the mechanism that causes neon lights to glow[5]. N C. 3 kV.")
23
The corona discharge replaces the electron filament in CI - the atmospheric pressure would quickly "burn out" any filaments - and produces primary N2°+ and N4°+ by electron ionisation. These primary ions collide with the vaporized solvent molecules to form secondary reactant gas ions - e.g. H3O+ and (H2O)nH+ (see fig. 2). These reactant gas ions then undergo repeated collisions with the analyte resulting in the formation of analyte ions. The high frequency of collisions results in a high ionisation efficiency and thermalisation of the analyte ions. This results in spectra of predominantly molecular species and adduct ions with very little fragmentation.
nH+ (see fig. 2). These reactant gas ions then undergo repeated collisions with the analyte resulting in the formation of analyte ions. The high frequency of collisions results in a high ionisation efficiency and thermalisation of the analyte ions. This results in spectra of predominantly molecular species and adduct ions with very little fragmentation..")
24
Atropin APCI tömegspektruma

25
MS-ANALIZÁTOROK Az analizátor választja el az ionforrásból nagy sebességgel érkező ionokat fajlagos tömegük szerint. Fajtái: 1. repülési idő (TOF: time of flight), 2. elektromos, pl. kvadrupól és ioncsapda, 3. mágneses analizátorú, 4. elektrosztatikus, 5. kettős fókuszálású (nagy felbotóképességű), 6. tandem (MS/MS, MSn) .
, 2. elektromos, pl. kvadrupól és ioncsapda, 3. mágneses analizátorú, 4. elektrosztatikus, 5. kettős fókuszálású (nagy felbotóképességű), 6. tandem (MS/MS, MSn) .")
26
TOF Ha minden töltéshordozó azonos kinetikus energiára tesz szert, akkor egyszeres iontöltés esetén: Z = iontöltése V = gyorsítófeszültség v = ion sebessége t = idő L = úthossz m = tömeg Ionok sebessége a tömegük négyzetgyökével fordítva arányos

27
Kalibrálás: pontosan ismert m/z értékű ionokra
Ha L = 1m és V = 2000V, akkor t N2+ = 8,37 μs és t O2+ = 8,94 μs

28
V/V0 = állandó ω a meghatározó!
A kvadrupólus tömeganalizátor négy párhuzamosan elhelyezkedő elektromosan vezető fémrúdból áll. Az ionforrásból érkező ionsugár a rudakkal párhuzamosan az általuk képezett üregen halad át. Az átlósan egymással szemben elhelyezkedő két-két rudat elektromos vezeték köti össze. Az így kialakult két rúd-pár szimultán kapcsolódik egy egyenáramú áramforrás ellentétes pólusaihoz és egy rádiófrekvenciás oszcillátorhoz. Az általuk létrehozott elektromos (kvadrupólus) tér a rudak között haladó ionsugárra merőleges. Az ionok az egyen- és váltóáram együttes hatására haladási irányukra merőleges transzverzális mozgást végeznek. A rudak közötti térben haladó pozitív ionokat az éppen pozitív töltésű rudak taszítják, míg a negatív töltésűek vonzzák. Mivel az oszcillátor hatására a fém rúd-párok relatív töltése folyamatosan változik az ionok szabálytalan oszcillációs mozgást végezve haladnak a rudak között. E mozgás az alkalmazott egyenáram Vdc feszültségétől, az oszcillátor frekvenciájától és amplitúdója Vac nagyságától, valamint az ionok m/z értékétől függ. A detektorba csak azok az ionok jutnak be, amelyek e mozgás során végig haladnak a négy rúd által határolt térben, a többi ion az elektródként viselkedő rudak valamelyikéhez csapódva elveszíti mozgási energiáját. A működés lényege, hogy a kvadrupól teret úgy változtatják, hogy a V/Vo állandó maradjon. Ezzel lényegében a tér frekvenciája változik. Csak az az ion képes az ionforrásból a detektorba eljutni, amelynek a sajátfrekvenciája azonos a kvadrupól tér pillanatnyi frekvenciájával.
 tér a rudak között haladó ionsugárra merőleges. Az ionok az egyen- és váltóáram együttes hatására haladási irányukra merőleges transzverzális mozgást végeznek. A rudak közötti térben haladó pozitív ionokat az éppen pozitív töltésű rudak taszítják, míg a negatív töltésűek vonzzák. Mivel az oszcillátor hatására a fém rúd-párok relatív töltése folyamatosan változik az ionok szabálytalan oszcillációs mozgást végezve haladnak a rudak között. E mozgás az alkalmazott egyenáram Vdc feszültségétől, az oszcillátor frekvenciájától és amplitúdója Vac nagyságától, valamint az ionok m/z értékétől függ. A detektorba csak azok az ionok jutnak be, amelyek e mozgás során végig haladnak a négy rúd által határolt térben, a többi ion az elektródként viselkedő rudak valamelyikéhez csapódva elveszíti mozgási energiáját. A működés lényege, hogy a kvadrupól teret úgy változtatják, hogy a V/Vo állandó maradjon. Ezzel lényegében a tér frekvenciája változik. Csak az az ion képes az ionforrásból a detektorba eljutni, amelynek a sajátfrekvenciája azonos a kvadrupól tér pillanatnyi frekvenciájával.")
29
Ioncsapda – Ion Trap (IT)
")
30
Az elektronemitterből érkező elektronok egy kapuelektródon át 50-80 eV-os energiával jutnak be az ioncsapda elektródok közé, ahová a mintát is bevezetjük. Ionizáció (EI, CI). Az ioncsapda elektródok olyan háromdimenziós teret hoznak létre, amelyben az ionok aperiodikus oszcillációra kényszerülnek, s a csapdában vannak mindaddig, amíg egy axiális amplitúdó moduláció (RF változtatása) az adott fajlagos tömegű és adott rezgésre képes iont az ionsokszorozó detektorba nem juttatja. Érzékenyebb, mint a kvadrupól. Kis helyigény. MS/MS könnyen megvalósítható.
 az adott fajlagos tömegű és adott rezgésre képes iont az ionsokszorozó detektorba nem juttatja. Érzékenyebb, mint a kvadrupól. Kis helyigény. MS/MS könnyen megvalósítható.")
31
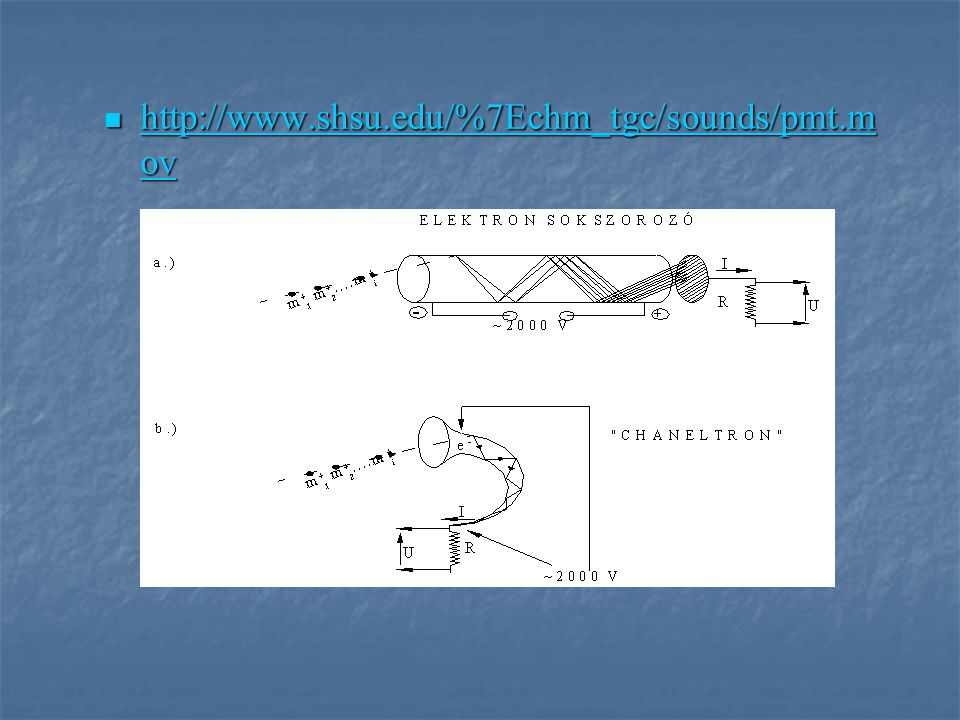
Detektor A detektor fő feladata az, hogy az egyes ionok számával arányos intenzitású jelet szolgáltasson. A legelterjedtebben ion-, vagy fotosokszorozó detektorokat használunk. Az ionsokszorozók (ionmultiplierek) eseté-ben a felfogó elektródra (dinód) becsapódó ionok elektronemissziót váltanak ki, ezek az elektronok a szemben elhelyezkedő elek-tródra csapódva szekunder elektronemisz-sziót hoznak létre ( jelerősítés).
 eseté-ben a felfogó elektródra (dinód) becsapódó ionok elektronemissziót váltanak ki, ezek az elektronok a szemben elhelyezkedő elek-tródra csapódva szekunder elektronemisz-sziót hoznak létre ( jelerősítés).")
33
Tömegspektrométerek jellemző adatai
1. felbontóképesség, 2. tömegtartomány, 3. felvételi sebesség, 4. kimutatási határ, 5. ionátviteli hatásfok, 6. hőmérséklettartomány. 2,Tömegtartomány: Da 4,A napi analitikai gyakorlatban a legtöbb készülékkel pg, vagy fg (femtogram) mennyiségek már megbízhatóan meghatározhatók. A TOF-MALDI ma a legkisebb mennyiségeket is kimutatni tudó analitikai mérőrendszer, amely lehetővé teszi akár g-nyi tömegű anyag kimutatását is. 5, .Ha "sok ion vész el" az ionforrás és a detektor között, akkor az "érzékenység" lecsökken. A kvadrupól és az ioncsapda tömegspektrométerek transzmissziója általában 70-80 %, míg a kettõs fókuszálású, "hosszú" tömegspektrométereké legfeljebb 40-50 %, amely a felbontóképesség növelésével tovább csökken. Analitikai szempontból azért is fontos a lehetõ legnagyobb transzmisszió, mert a tömegspektrométerbe bekerülõ molekulák ionizációjának hatásfoka (az EI ionforrásban) általában 10 % körüli. 6, A sokféle lehetséges meg-fontolás mellett az a döntő, hogy a vizsgálandó alkotónak az ionizáció bekövetkeztéig gáz fázisban kell maradnia. (Az EI ionforrás ún. "gázionforrás".) Ezt a legtöbb készülék ionforrása C, illetve C között biztosítani tudja.
 mennyiségek már megbízhatóan meghatározhatók. A. TOF-MALDI ma a legkisebb mennyiségeket is kimutatni tudó analitikai mérőrendszer, amely lehetővé teszi akár g-nyi tömegű anyag kimutatását is. 5, .Ha sok ion vész el az ionforrás és a detektor között, akkor az érzékenység lecsökken. A kvadrupól és az ioncsapda tömegspektrométerek transzmissziója általában %, míg a kettõs fókuszálású, hosszú tömegspektrométereké legfeljebb %, amely a felbontóképesség növelésével tovább csökken. Analitikai szempontból azért is fontos a lehetõ legnagyobb transzmisszió, mert a tömegspektrométerbe bekerülõ molekulák ionizációjának hatásfoka (az EI ionforrásban) általában 10 % körüli. 6, A sokféle lehetséges meg-fontolás mellett az a döntő, hogy a vizsgálandó alkotónak az ionizáció bekövetkeztéig gáz fázisban kell maradnia. (Az EI ionforrás ún. gázionforrás .) Ezt a legtöbb készülék ionforrása C, illetve C között biztosítani tudja.")
34
A felbontóképesség: adott tömegtartományban két egymás melletti, eltérő tömegű ion mennyire különböztethető meg egymástól. Azokat a készülékeket, amelyek felbontóképessége Rs>104, nagy felbontóképességű, amelyeké Rs<104, kis felbontóképességű tömegspektrométereknek nevezzük. A nagy felbontóképességű készülékek mind fõként kettõs fókuszálású tömegspektrométerek és ezek szerkezetvizsgálatot tesznek lehetõvé. Ehhez ugyanis a megbízható elem-összetétel ismerete elengedhetetlen, ezt pedig legalább a tömeg második tizedes jegyének a pontos ismeretében van csak módunk megbízhatóan kiszámítani. A kis felbontóképességû (Rs<104) készülékek többnyire felbontóképességgel rendelkeznek. Ez azt jelenti, hogy Rs=1000 esetén m=100 mellett m=0,1, azaz ekkora tömegkülönbség még egyértelműen mérhetõ. A karakterisztikus tömegspektrum felvételéhez ennél nagyobb Rs nem is szükséges, hiszen a szerves molekulákban a legkisebb elemi tömegegység különbség mindig legalább 1-hez közeli (a H tömege 1,007892) érték, vagy ennél nagyobb. Az analitikai készülékek mind kis felbontóképességűek. Így a kvadrupól MS felbontóképessége , az ioncsapdáé ( A TOF készülékeké is ).
 készülékek többnyire felbontóképességgel rendelkeznek. Ez azt jelenti, hogy Rs=1000 esetén m=100 mellett m=0,1, azaz ekkora tömegkülönbség még egyértelműen mérhetõ. A karakterisztikus tömegspektrum felvételéhez ennél nagyobb Rs nem is szükséges, hiszen a szerves molekulákban a legkisebb elemi tömegegység különbség mindig legalább 1-hez közeli (a H tömege 1,007892) érték, vagy ennél nagyobb. Az analitikai készülékek mind kis felbontóképességűek. Így a kvadrupól MS felbontóképessége , az ioncsapdáé ( A TOF készülékeké is ).")
35
Tömegspektrometria alkalmazása
gázelemzés: lámpa töltőgázok elemzése, fermen-tációs gázelegyek. izotóparány mérés: kőzetek, ásványok, biológiai rendszerek elemeinek izotóparány meghatá-rozása (pl. geológiai kormeghatározás, fossziliák kora). szervetlen környezetszennyezők elemzése: ICP-MS. szerves szerkezetvizsgálat (pontos tömegmérés, elemösszetétel, szerkezet meghatározása céljá-ból). szerves rendszerek minőségi és mennyiségi összetételének meghatározása (GC-MS, LC-MS).
. szervetlen környezetszennyezők elemzése: ICP-MS. szerves szerkezetvizsgálat (pontos tömegmérés, elemösszetétel, szerkezet meghatározása céljá-ból). szerves rendszerek minőségi és mennyiségi összetételének meghatározása (GC-MS, LC-MS).")
36
A mintaelőkészítés műveletei
1, Fizikai műveletek: - mintavétel (homogén, heterogén, statisztikus, vagy random kiválasztás), - aprítás, őrlés, homogenizálás, - oldat készítés, - szitálás, szűrés, dialízis, - centrifugálás és ultracentrifugálás, - betöményítés és szárítás (vakum bepárlás és fagyasztva szárítás - liofilizálás), - kivonás (extrakció). 2, Kémiai műveletek: - hígítás, pH beállítása, - komplexképző adalékok. - származékkészítés.
, - aprítás, őrlés, homogenizálás, - oldat készítés, - szitálás, szűrés, dialízis, - centrifugálás és ultracentrifugálás, - betöményítés és szárítás (vakum bepárlás és fagyasztva szárítás - liofilizálás), - kivonás (extrakció). 2, Kémiai műveletek: - hígítás, pH beállítása, - komplexképző adalékok. - származékkészítés.")
37
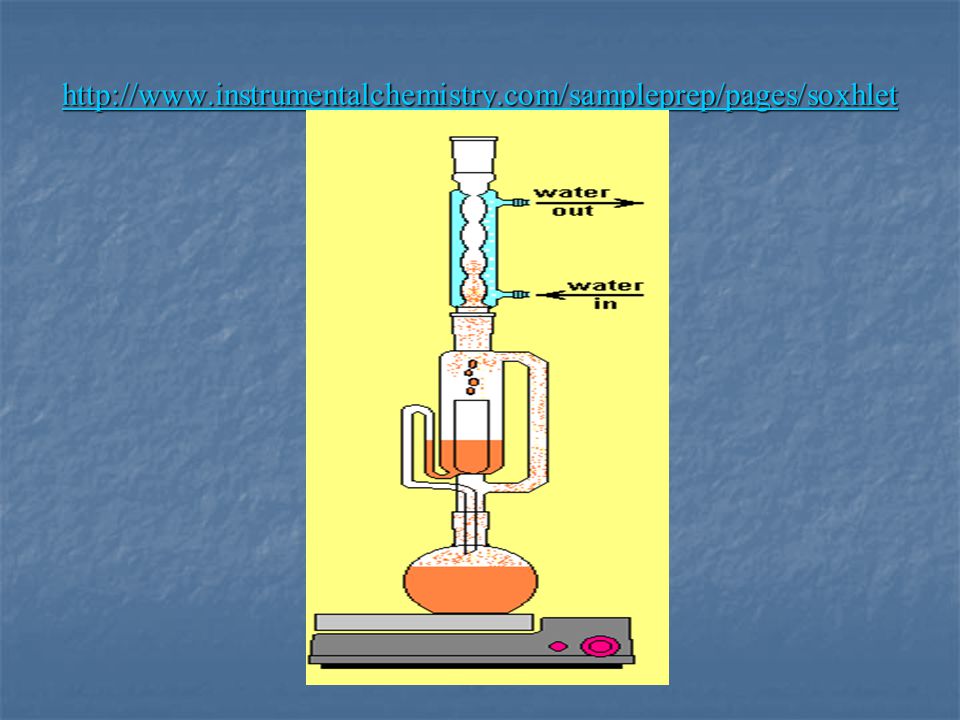
Szilárd-folyadék extrakció
1, Soxhlet extrakció Szilárd minták szerves anyag tartalmának klasszikus extrakciós módszere . Nem, vagy félig illékony összetevők meghatá-rozására. A mintát porózus cellulóz csőbe helyezik. Jól oldó oldószert áramoltatnak a mintán keresztül. A cirkuláció melegítés hatására történik. Idő és oldószerigénye igen nagy (16-24 óra).
.")
38
Lipidek kivonása tejből
A fűtött lombikból felszálló oldószergőzök a hűtőfeltétről visszacsepegnek a szilárd mintára. Miután a felső részben az oldószer feltölti a jobb oldalon levő közlekedő edényt is, az oldószer visszafolyik az alsó lombikba és a ciklus kezdődik elölről. Szilárd minták esetén a módszer kimerítő extrakciót tesz lehetővé, de igen hosszadalmas. A körülményektől függően az extrakció időtartama 8-48 óra. A módszer automatizálható, a kereskedelemben kaphatók olyan berendezések, amelyekkel több minta párhuzamosan extrahálható. SOXHLET Franz Lipidek kivonása tejből

40
2, Automata Soxhlet extrakció
Az oldószer és a minta folyamatos és közvetlen érintkezésének köszönhetően jelentősen csökken az idő és az oldószer szükséglet. Az extrakciót az oldószer forráspontján végzik. Lépések (2 óra): - minta és az oldószer érintkeztetése - minta és az oldószer elválasztása az extraktum betöményítése 1-2 ml-re. Hátránya, hogy az alacsony forráspontú összete-vők egy része a bepárlásnál elvész.
: - minta és az oldószer érintkeztetése. - minta és az oldószer elválasztása. az extraktum betöményítése 1-2 ml-re. Hátránya, hogy az alacsony forráspontú összete-vők egy része a bepárlásnál elvész.")
41
Gyorsított oldószeres extrakció Accelerated Solvent Extraction (ASE)
Gyorsított oldószeres extrakció (EPA Method 3545A) A használt ASE rövidítés az angol Accelerated Solvent Extraction elnevezésből ered. Szintén műszeres módszer, ahol szabályozott, emelt nyomás és hőmérséklet alkalmazásával növelik meg az extrakció hatásfokát. A megfelelő módszerek jelenleg is kidolgozás alatt állnak, de alkalmazása egyre gyakoribb, a kutatás mellett a gyakorlatban is. A módszer félautomata, de jelentős az idő és az oldószer szükséglet csökkenése. A cella töltése után az extrakció, elválasztás és gyűjtési lépés automatikus. A berendezésnek vannak olyan változatai is melyek akár 6-12 minta egyidejű extrakcióját teszik lehetővé [37]. Az extrakciós folyamat az oldószer atmoszférikus forráspontjának két-háromszorosának megfelelő hőmérsékleten történik, ami a nyomás szabályozásával érhető el. A minta és az oldószer érintkeztetési 5-10 perc. Az extraktum ezt követően a gyűjtő edénybe kerül. A kis oldószer/minta tömegarány miatt a mintákat közvetlenül gázkromatográfiásan mérni lehet.
 A használt ASE rövidítés az angol Accelerated Solvent Extraction elnevezésből ered. Szintén műszeres módszer, ahol szabályozott, emelt nyomás és hőmérséklet alkalmazásával növelik meg az extrakció hatásfokát. A megfelelő módszerek jelenleg is kidolgozás alatt állnak, de alkalmazása egyre gyakoribb, a kutatás mellett a gyakorlatban is. A módszer félautomata, de jelentős az idő és az oldószer szükséglet csökkenése. A cella töltése után az extrakció, elválasztás és gyűjtési lépés automatikus. A berendezésnek vannak olyan változatai is melyek akár 6-12 minta egyidejű extrakcióját teszik lehetővé [37]. Az extrakciós folyamat az oldószer atmoszférikus forráspontjának két-háromszorosának megfelelő hőmérsékleten történik, ami a nyomás szabályozásával érhető el. A minta és az oldószer érintkeztetési 5-10 perc. Az extraktum ezt követően a gyűjtő edénybe kerül. A kis oldószer/minta tömegarány miatt a mintákat közvetlenül gázkromatográfiásan mérni lehet.")
42
Foszfor- és klórtartalmú peszticidek és herbicidek
Alkalmazása: Vízoldhatatlan, vagy vízben kevéssé oldódó szerves anyagok extrakciója szilárd mintákból (föld, agyag, üledék, iszap, stb.) Foszfor- és klórtartalmú peszticidek és herbicidek Poliklorozott bifenilek (PCB) Poliklorozott dibenzodioxidok és dibenzofuránok. Hőmérséklet: 100–180 °C Nyomás: 1500–2000 psi Előnyei: Kismennyiségű oldószer (pár ml) Rövid idő (10-20 perc) Nevertheless, the PFE technique is now well accepted as an alternative to Soxhlet extraction. EPA Method 3545A, entitled "Pressurized Fluid Extraction," is a recognized alternative procedure for extracting water-insoluble or slightly water-soluble organic compounds from soils, clays, sediments, sludge, and waste solids. This method is applicable to the extraction of semivolatile organic compounds, organophosphorus pesticides, organochlorine pesticides, chlorinated herbicides, polychlorinated biphenyls (PCBs), and polychlorinated dibenzodioxins and dibenzofurans (PCDDs and PCDFs), which can then be analyzed by a variety of chromatographic procedures. PFE, sometimes referred to as pressurized solvent extraction (PSE), uses elevated temperatures (100–180 °C) and pressures (1500–2000 psi) to achieve analyte recoveries equivalent to those from Soxhlet extraction but using less solvent and taking significantly less time. The idea is that a solvent heated above its boiling point in a closed system becomes a very potent extraction solvent. Extractions by PFE generally require only 10–20 min. Sample requirements vary but generally 1–30 g is required depending upon the sensitivity needed. Sample extraction cells can be as small as 1 mL or as large as 100 mL.
 Foszfor- és klórtartalmú peszticidek és herbicidek. Poliklorozott bifenilek (PCB) Poliklorozott dibenzodioxidok és dibenzofuránok. Hőmérséklet: 100–180 °C. Nyomás: 1500–2000 psi. Előnyei: Kismennyiségű oldószer (pár ml) Rövid idő (10-20 perc) Nevertheless, the PFE technique is now well accepted as an alternative to Soxhlet extraction. EPA Method 3545A, entitled Pressurized Fluid Extraction, is a recognized alternative procedure for extracting water-insoluble or slightly water-soluble organic compounds from soils, clays, sediments, sludge, and waste solids. This method is applicable to the extraction of semivolatile organic compounds, organophosphorus pesticides, organochlorine pesticides, chlorinated herbicides, polychlorinated biphenyls (PCBs), and polychlorinated dibenzodioxins and dibenzofurans (PCDDs and PCDFs), which can then be analyzed by a variety of chromatographic procedures. PFE, sometimes referred to as pressurized solvent extraction (PSE), uses elevated temperatures (100–180 °C) and pressures (1500–2000 psi) to achieve analyte recoveries equivalent to those from Soxhlet extraction but using less solvent and taking significantly less time. The idea is that a solvent heated above its boiling point in a closed system becomes a very potent extraction solvent. Extractions by PFE generally require only 10–20 min. Sample requirements vary but generally 1–30 g is required depending upon the sensitivity needed. Sample extraction cells can be as small as 1 mL or as large as 100 mL.")
43
Felhasznált oldószer mennyisége Mikrohullámú feltárás
Technika Felhasznált oldószer mennyisége Soxhlet ml Automata soxhlet ml Szonikálás ml Mikrohullámú feltárás 25-50 ml ASE 15-45 ml

44
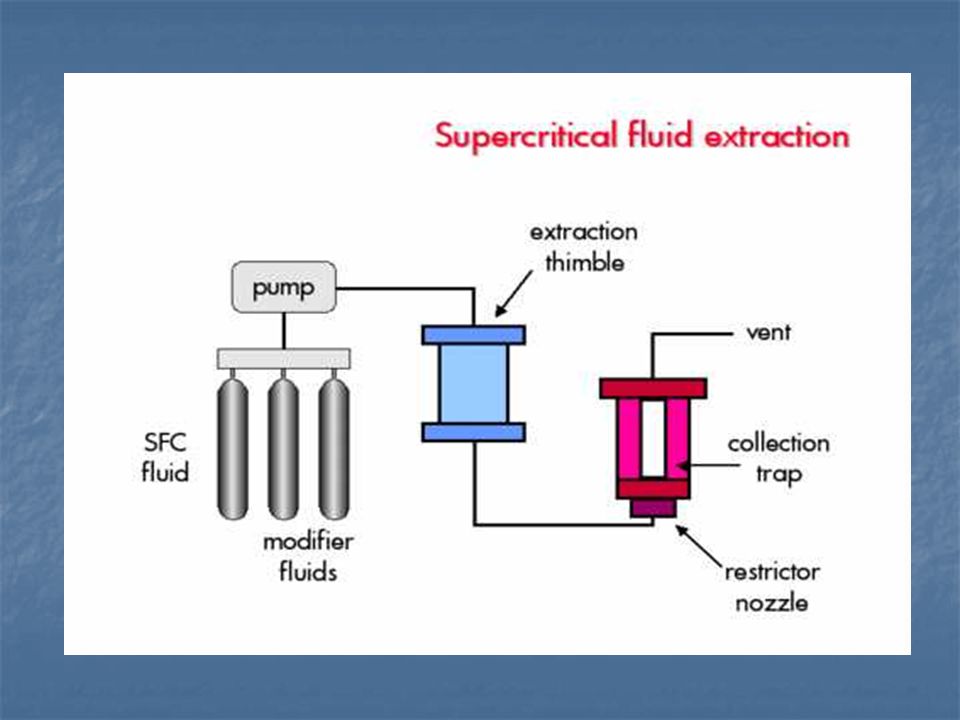
Szuperkritikus folyadék extrakció - 1990
Extraháló oldószer: szuperkritikus állapotú széndioxid (~ 70 bar, 30ºC). A CO2 apoláris jellegű (~ toluol, etil-acetát), ezért ha például PCB-ket akarunk kinyerni, akkor segédoldószert kell alkalmazni. Segédoldószerek: - a klórozott szénhidrogének (CH2Cl2, CHCl3), tetrahidrofurán, izopropanol. Egy egykomponensű anyag fázisdiagramja megadja azokat a hőmérséklet- és nyomástartományokat, amelyeknél a különböző fázisok termodinamikailag stabilak. A tartományok közötti határvonalak megadják azokat a p és T értékeket, amelyek mentén két fázis dinamikus egyensúlyban van. A CO2 fázisdiagramján (9. ábra) látható, hogy egy adott hőmérsékleten (Tc) eltűnik a folyadék/gőz fázishatár. A Tc érték felett az anyag semmilyen nyomáson nem cseppfolyósítható. CO2 fázisdiagramja
. A CO2 apoláris jellegű. (~ toluol, etil-acetát), ezért ha például PCB-ket akarunk kinyerni, akkor segédoldószert kell alkalmazni. Segédoldószerek: - a klórozott szénhidrogének (CH2Cl2, CHCl3), tetrahidrofurán, izopropanol. Egy egykomponensű anyag fázisdiagramja megadja azokat a hőmérséklet- és nyomástartományokat, amelyeknél a különböző fázisok termodinamikailag stabilak. A tartományok közötti határvonalak megadják azokat a p és T értékeket, amelyek mentén két fázis dinamikus egyensúlyban van. A CO2 fázisdiagramján (9. ábra) látható, hogy egy adott hőmérsékleten (Tc) eltűnik a folyadék/gőz fázishatár. A Tc érték felett az anyag semmilyen nyomáson nem cseppfolyósítható. CO2 fázisdiagramja.")
45
Szuperkritikus közeg egyszerre viselkedik gázként és folyadékként.
A gáz, a folyadék, illetve a szuperkritikus állapot jellemzői Tulajdonság Mérték egység Gáz Folyadék Szuperkriti-kus fluid Sűrűség () g/cm3 10-3 1 0,3 Diffúziós állandó (Dm) cm-2/s 10-1 510-6 Viszkozitás () g/(cms) 10-4 10-2
 g/cm ,3. Diffúziós állandó (Dm) cm-2/s 10-6. Viszkozitás () g/(cms)")
46
A folyamatot befolyásoló elsődleges paramé-terek:
- a széndioxid és a segédoldószer aránya T p Előnye a módszernek, hogy az oldószer eltávolítása a nyomás megszüntetésével egyszerűen megoldható és csak az extraktum marad vissza. Környezetbarát oldószer. Nehéz standardizálni és költséges a berendezés.

48
Klasszikus folyadék-folyadék extrakció

49
A folyadék-folyadék extrakciót vizes közegű mintáknál alkalmazzuk
A folyadék-folyadék extrakciót vizes közegű mintáknál alkalmazzuk. Az eljárás választó-tölcsérben való kirázást jelent, a meghatározni kívánt összetevőnek megfelelő, vízzel nem elegyedő oldószerrel. (K - megoszlási hányados). Apoláris oldószerek : hexán, heptán Halogéntartalmú oldószerek: diklórmetán, szén-tetraklorid (toxicitás!). Poláris oldószer: etil-acetát Savak – alacsony pH-n Bázikus vegyületek – magas pH-n
. Apoláris oldószerek : hexán, heptán. Halogéntartalmú oldószerek: diklórmetán, szén-tetraklorid (toxicitás!). Poláris oldószer: etil-acetát. Savak – alacsony pH-n. Bázikus vegyületek – magas pH-n.")
50
Szilárd fázisú extrakció – Solide Phase Extraction (SPE)
")
51
Lépések

52
SPE töltetek Fordított fázisú C18, C8, C6, C4, C2 Normál fázisú
Szilikagél, aluminium-oxid, diol, amino, ciano, fenil Ioncserés SAX, SCX Polimer gyanták Sztirol-divinil-benzol kopolimer Oasis HLB Vegyes Fordított fázis + ioncserés, Oasis MCX, Oasis MAX Aminopropyl [NH2]Silica-based, moderately polar, bonded phase with weakly basic surface; can be used as a polar sorbent, like silica, with different selectivity for acidic/basic analytes or as weak anion exchanger in aqueous medium below pH 8.Applications include phenols and phenolic pigments, petroleum fractionation, saccharides, drugs and drug metabolites Cyanopropyl [CN]Silica-based bonded phase of low hydrophobicity; can be used as less polar alternative to silica in normal-phase applications or as less hydrophobic alternative to C18 or C8 in reversed-phase applications.Typical applications include drugs, drug metabolites, and pesticides. DiolSilica-based, moderately polar, bonded phase with neutral surface; can be used as an alternative to silica in normal phase applications, where the acidic character of silica is undesirable or as very weakly interacting hydrophobic phase in aqueous media.Applications include antibiotics from cosmetics; isolation of proteins or peptides by hydrophobic interaction chromatography.

53
Oasis® HLB (hydrophilic-lipophilic-balance)
3x a kapacitása a C18-nak Könnyen nedvesedik, nem szárad ki, mint a szilika, jobb a reprodukálhatósága. 1-vinil-2-pirrolidinon Pore Size (nominal): 80 Å Particle Size: 30 µm [or 60 µm for LP grade] Surface Functionality: m-Divinylbenzene & N-vinylpyrrolidone copolymer
: 80 Å. Particle Size: 30 µm [or 60 µm for LP grade] Surface Functionality: m-Divinylbenzene & N-vinylpyrrolidone copolymer.")
54
1, Töltet tömeg (bed mass) Szilika – töltet tömeg 5%-a a kapacítása
(100 mg – 5 mg anyagot köt). Polimer töltetek – nagyobb felület → nagyobb kapacitás (2-3x) → 10-15% 2, Töltet térfogat = pórusok közötti tér + pórustérfogat Kondicionálás, mosás és eluálás térfogata = 4-8 X töltet térfogatnak Szilika – 150 μl/100mg ~ 1,2 ml Polimer – 250 μl/100mg ~ 2 ml As a general rule for selecting the proper bed mass Silica-based sorbents: Non-ionic sorbents retain a mass of solute (analyte plus retained contaminants) that is equivalent to approximately 5% of the sorbent mass. Therefore, a 100mg cartridge can retain approximately 5mg of total solute mass. Polymeric sorbents The large surface area of polymers give a higher capacity that is approximately 10-15% of the sorbent mass. In general, solvent volumes between 4 to 8 times the bed volume are necessary to ensure proper conditioning, washing and elution. Conditioning with less than 4 to 8 bed volumes increases the risk of incomplete solvation of the bed and low or irreproducible recoveries, while more than 4 bed volumes is typically unnecessary. Conventional packed bed, silica-based SPE products typically have a bed volume of approximately 150µL per 100mg of sorbent. Polymeric sorbents require larger solvent volumes for proper conditioning, washing and eluting. It is recommended to use µL per 100mg of sorbent.
. Polimer töltetek – nagyobb felület → nagyobb kapacitás (2-3x) → 10-15% 2, Töltet térfogat = pórusok közötti tér + pórustérfogat. Kondicionálás, mosás és eluálás térfogata = 4-8 X töltet térfogatnak. Szilika – 150 μl/100mg ~ 1,2 ml. Polimer – 250 μl/100mg ~ 2 ml. As a general rule for selecting the proper bed mass Silica-based sorbents: Non-ionic sorbents retain a mass of solute (analyte plus retained contaminants) that is equivalent to approximately 5% of the sorbent mass. Therefore, a 100mg cartridge can retain approximately 5mg of total solute mass. Polymeric sorbents. The large surface area of polymers give a higher capacity that is approximately 10-15% of the sorbent mass. In general, solvent volumes between 4 to 8 times the bed volume are necessary to ensure proper conditioning, washing and elution. Conditioning with less than 4 to 8 bed volumes increases the risk of incomplete solvation of the bed and low or irreproducible recoveries, while more than 4 bed volumes is typically unnecessary. Conventional packed bed, silica-based SPE products typically have a bed volume of approximately 150µL per 100mg of sorbent. Polymeric sorbents require larger solvent volumes for proper conditioning, washing and eluting. It is recommended to use µL per 100mg of sorbent.")
55
Molecularly imprinted polymers
MIPs are highly cross-linked polymer phases that have pre-determined selectivity for a single analyte or a group of structurally related analytes. The SupelMIP SPE products consist of highly cross-linked polymers that are engineered to extract a single analyte of interest or a class of structurally related analytes of interest with an extremely high degree of selectivity. This is possible because selectivity is introduced during MIP synthesis in which a template molecule, designed to mimic the analyte, guides the formation of specific cavities or imprints that are sterically and chemically complementary to the analyte(s) of interest. The Benefits of SupelMIP SPE · Achieve lower detection limits through superior selectivity · Save time and reduce cost via robust and rapid methodology · Stable at broad pH ranges and high temperatures · Reduce ion suppression · Stringent quality control conditions MIP binding site is both chemically and sterically complementary to the analyte(s) of interest. Multiple non-covalent interaction points (ion-exchange, reversed-phase with polymer backbone, and hydrogen bonding) between the MIP phase and analyte functional groups allow for stronger analyte retention. Improved selectivity is then introduced through the use of harsher wash conditions during sample prep methodology.
 of interest. The Benefits of SupelMIP SPE · Achieve lower detection limits through superior selectivity · Save time and reduce cost via robust and rapid methodology · Stable at broad pH ranges and high temperatures · Reduce ion suppression · Stringent quality control conditions MIP binding site is both chemically and sterically complementary to the analyte(s) of interest. Multiple non-covalent interaction points (ion-exchange, reversed-phase with polymer backbone, and hydrogen bonding) between the MIP phase and analyte functional groups allow for stronger analyte retention. Improved selectivity is then introduced through the use of harsher wash conditions during sample prep methodology.")
56
Amphetamine related drugs are currently among the most wellknown
drugs of abuse in sports, in the workplace and by recreational users. Screening and confirmative analysis is done by forensic, clinical and doping laboratories.

57
Micro Extraction by Packed Sorbent (MEPS)
")
59
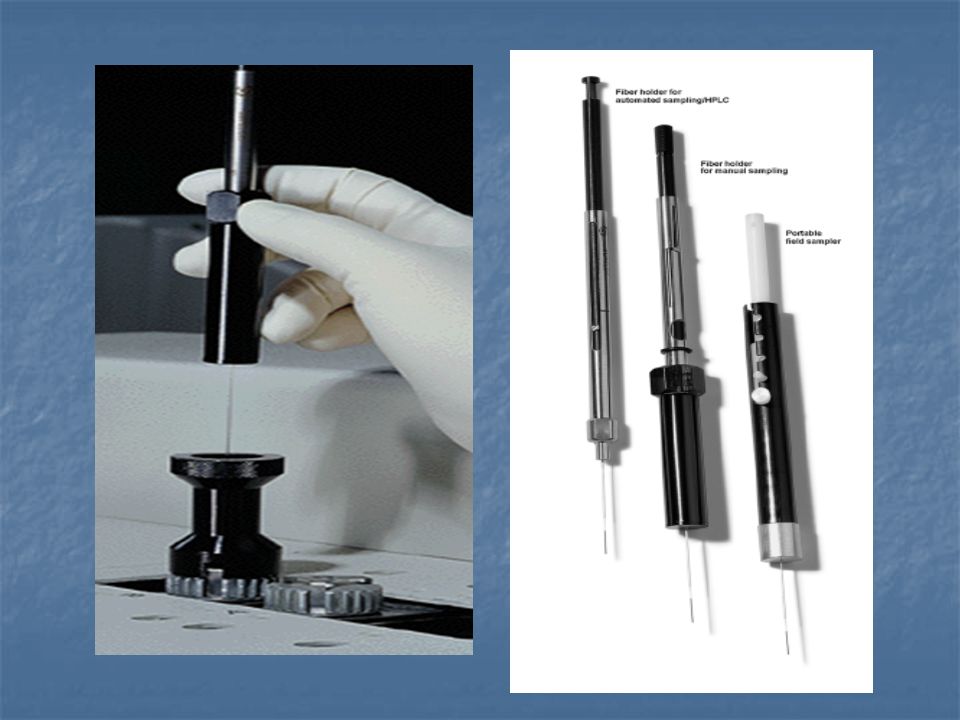
Szilárd fázisú mikroextrakció - Solide Phase Microextraction (SPME)
")
61
Az olvasztott kvarcszálat (fiber), a vízmintába meríttetik állandó keverés mellett, ekkor a szerves mikroszennyezők abszorbeálódnak az adott komponensre jellemző megosz-lási hányados értékének megfelelően. Időtartama perc. A fiberen szerves folyadékfilm van.

62
A mintát tartalmazó kvarcszálat a gázkromatográf fűtött injektorába juttatva ( C) a szerves komponensek deszorbeálódnak.
 a szerves komponensek deszorbeálódnak.")
63
Alkalmazott folyadékfilmek
1, Polidimetil-sziloxán (PDMS) 2, Polidimetil-sziloxán /Divinil-benzol (PDMS/DVB) 3, Carbowax/ Divinil-benzol (CW/DVB) 4, Poliakrilát (PA)
 2, Polidimetil-sziloxán /Divinil-benzol (PDMS/DVB) 3, Carbowax/ Divinil-benzol (CW/DVB) 4, Poliakrilát (PA)")
64
< TARGET="display">

65
Gőztér analízis (Head Space)
")
66
Hőmérséklet hatása a megoszlásra

67
http://www. entechinst

68
Purge and Trap Illékony apoláris szerves komponensek vízmintá-ból történő extrakciójára használják. 5-20 ml vízmintán nagytisztaságú gáz (pl. He) buborékol keresztül. Az illékony szerves komponensek a gázbuborékokkal eltávoznak és egy alkalmas adszorbensen szobahőmérsékleten adszorbeálódnak. A fűthető adszorbens oszlopról a minta néhány cm-es hűtött kapillárisba kerül, ahol lecsapódik. Ez a kriofókuszálás. Deszorpció: néhány másodperc alatt C -ra felfűtik a kapillárist és a minta GC-be kerül.
 buborékol keresztül. Az illékony szerves komponensek a gázbuborékokkal eltávoznak és egy alkalmas adszorbensen szobahőmérsékleten adszorbeálódnak. A fűthető adszorbens oszlopról a minta néhány cm-es hűtött kapillárisba kerül, ahol lecsapódik. Ez a kriofókuszálás. Deszorpció: néhány másodperc alatt C -ra felfűtik a kapillárist és a minta GC-be kerül.")
70
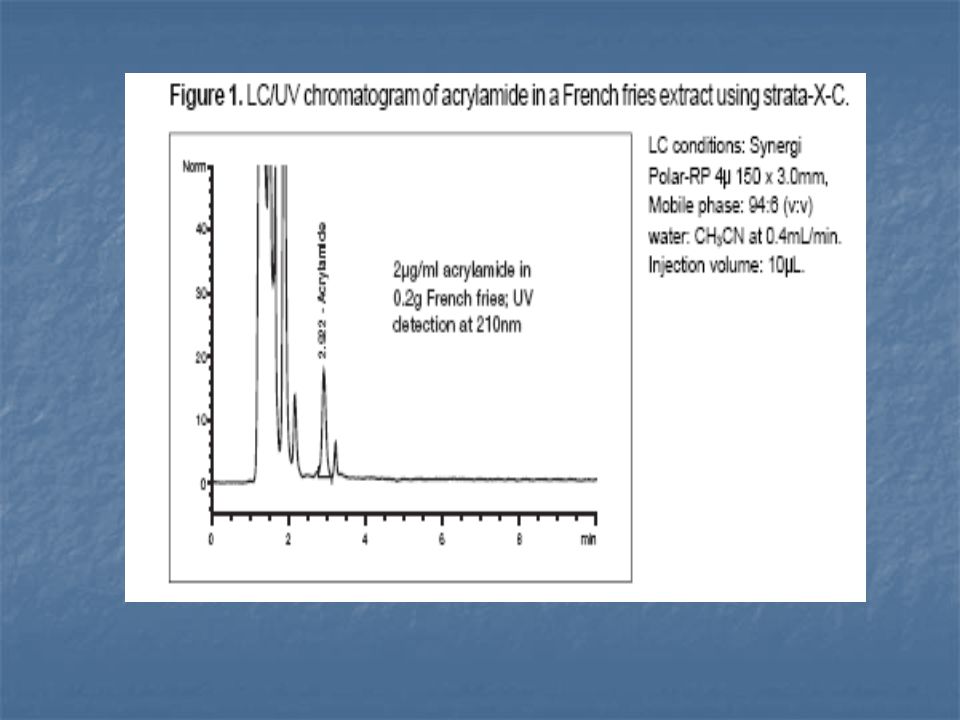
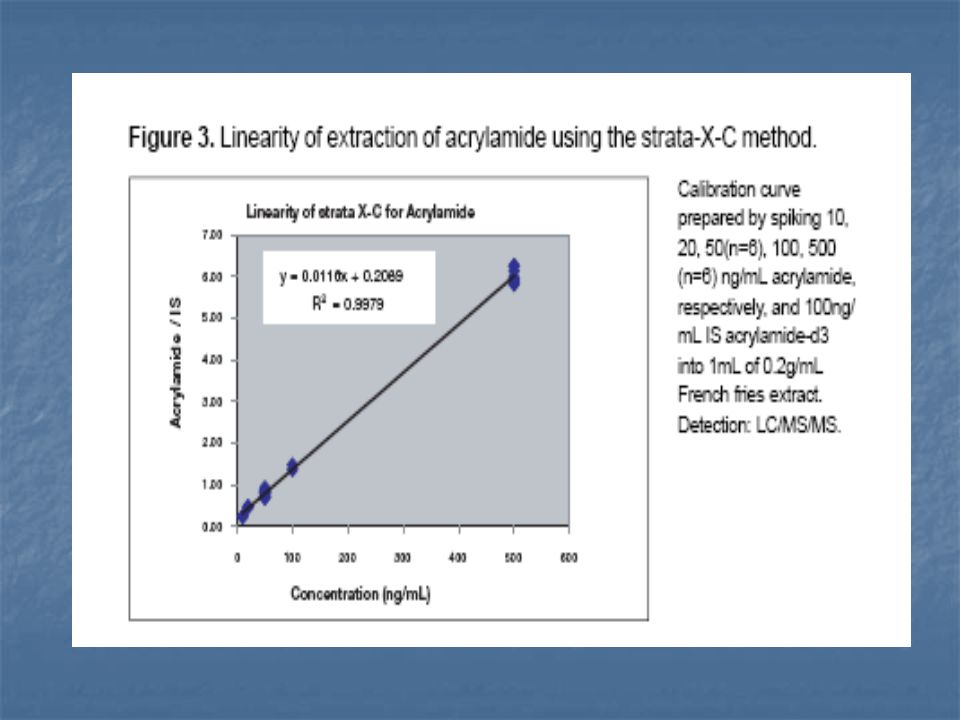
A legújabb vizsgálatok arra utalnak, hogy redukáló cukrok jelenlétében alacsony víztartalom mellett aszparaginból hevítés hatására keletkezik az akrilamid.

72
Protonált molekulaion, amiből elmegy az ammónia

74
Mérési eredmény = valódi érték + rendszeres hiba + véletlen hiba
Valódi értéket sosem ismerjük tökéletesen pontosan, véletlenszerű hibákat valószínűségi folyamatok eredményének tekintjük leírásukra a valószínűségszámítást használjuk. Mérési eredmény = valódi érték + rendszeres hiba + véletlen hiba Valódi érték???????

75
A mérési adatok eloszlási diagramja és az elméleti Gauss-féle elosztást reprezentáló görbe
Ha a mérések számát a végtelenségig növeljük, az intervallumokat pedig ezzel párhuzamosan csökkentjük, akkor egy harang alakú eloszlási görbét kapunk, amelyet Gauss, vagy normál eloszlási görbének nevezünk.

76
A mérési adatok szóródását az átlag érték körül a standard deviáció (σ) és négyzete a variancia (σ2) írja le. Véges számú mérést tudunk végezni → becslést alkalmazunk RSD%

77
Visszanyerés Mérendő komponens mennyiségét ismert mértékben megnövelik (spiking) és az adott analitikai módszerrel megmérik az eredeti és a megnövelt koncentrációjú mintát. A visszanyerés a két mért koncentráció különbségének és a valóságos növekménynek az aránya. Ha a koncentráció növekményre nézve a visszanyerés gyenge, a módszer jelentősen torzít.

Hasonló előadás
















>")




