Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
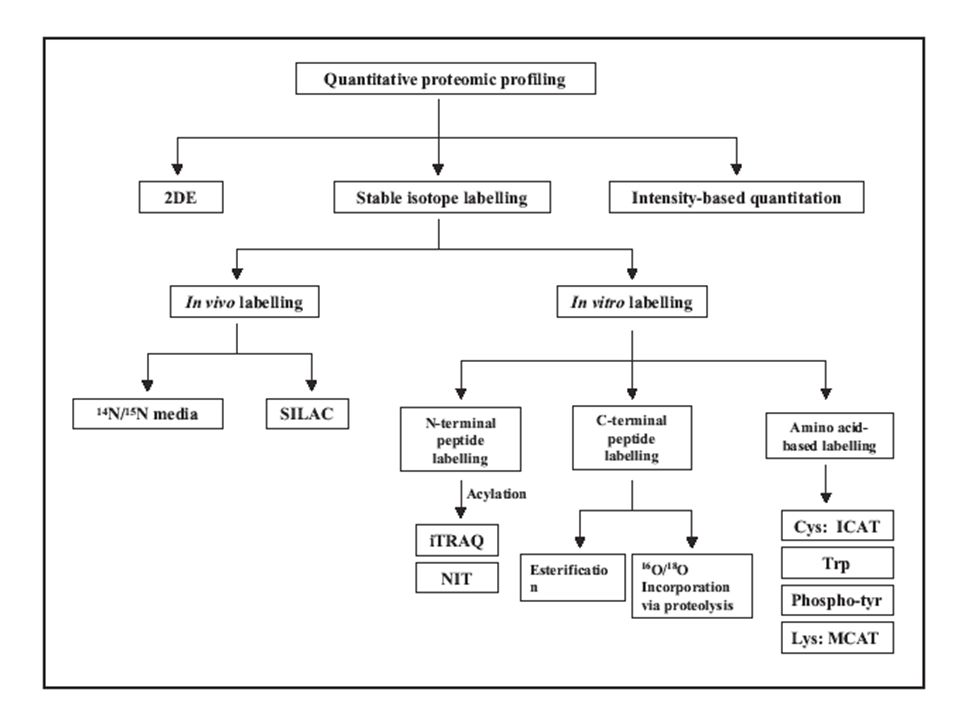
Eddig csak kvali volt... Kvantitatív proteomika 1) a frakcionálás szintjén Pl. 2D gélek összehasonlítása vizuálisan, komputer programokkal, differenciál festéssel 2) az MS-analízis során
 az MS-analízis során.")
3
Az MS NEM kvantitatív módszer * a különböző molekulák nem egyformán detektálhatók pl. különböző peptidek relatív intenzitása nem tükrözi relatív mennyiségüket * nem minden komponenst detektálunk az elegyekből

4
Hogy lehet ezt legyőzni? csak nagyon hasonló peptidek relatív intenzitását vethetjük össze! 1) Jelöljünk két sejt-populációt hasonlóan, de megkülönböztethetően! 2) Keverjük össze az összes fehérjét! 3) A keveréket emésszük, analizáljuk! És így kvantizhatunk!
 Jelöljünk két sejt-populációt hasonlóan, de megkülönböztethetően. 2) Keverjük össze az összes fehérjét. 3) A keveréket emésszük, analizáljuk. És így kvantizhatunk!.")
5
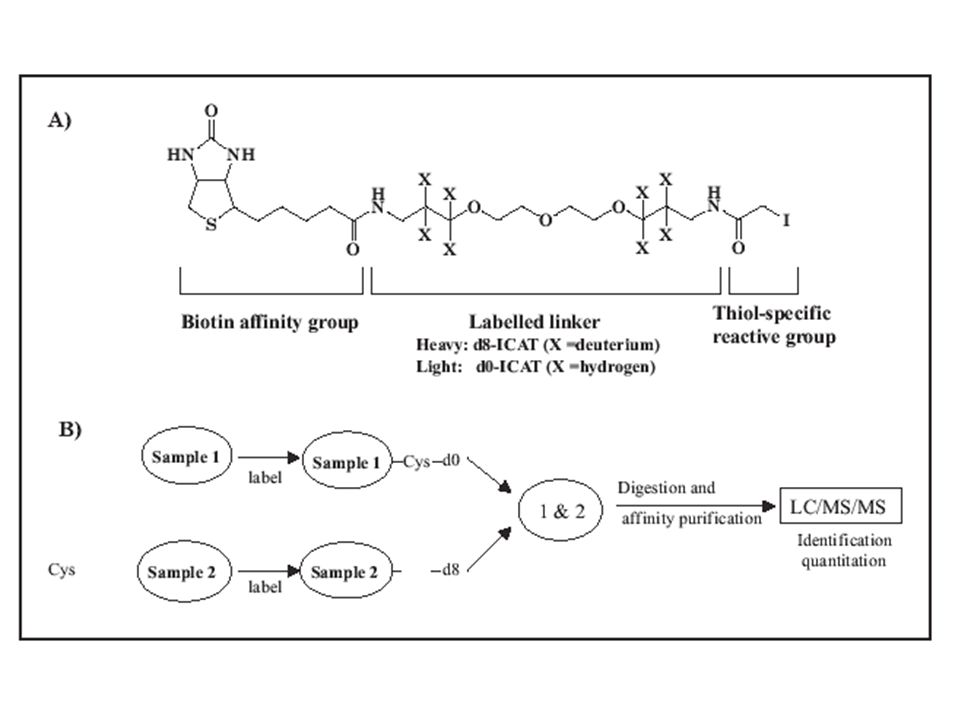
Hogyan jelölhetünk? ICAT (Isotope-Coded Affinity Tags) – Cys-oldalláncának alkilezése, könnyű (H 8 ) és nehéz (D 8 ) alkil-csoporttal → amiben nincs Cys, az kiesik → a deuterált vegyület retenciós ideje kicsit hosszabb azonosítás: MS-MS alapon kvanti: MS-alapon relatív intenzitásokból ingadozás: van az 30% is!
 – Cys-oldalláncának alkilezése, könnyű (H 8 ) és nehéz (D 8 ) alkil-csoporttal → amiben nincs Cys, az kiesik → a deuterált vegyület retenciós ideje kicsit hosszabb azonosítás: MS-MS alapon kvanti: MS-alapon relatív intenzitásokból ingadozás: van az 30% is!.")
7
Hogyan jelölhetünk? SILAC (Stable Isotope Labeling with amino acids in Cell Cultures) C 13, N 15, O 18 tápanyaggal visszük be – jelölt aminosav → Olyat kell választani, ami nem alakulhat át más aminosavvá, illetve nem szintetizálódik a sejtben Tripszines emésztéshez Arg a menő! a keveréket frakcionáljuk, emésztjük, analizáljuk azonosítás MS-MS alapon kvanti a relatív intenzitásból
 C 13, N 15, O 18 tápanyaggal visszük be – jelölt aminosav → Olyat kell választani, ami nem alakulhat át más aminosavvá, illetve nem szintetizálódik a sejtben Tripszines emésztéshez Arg a menő. a keveréket frakcionáljuk, emésztjük, analizáljuk azonosítás MS-MS alapon kvanti a relatív intenzitásból.")
8
Hogyan jelölhetünk? tripszines emésztés H 2 O 18 -ban Két oxigén lép be, minden peptid jelölődik Rengeteg információ Még a szekvenálás is könnyebb, a C- terminális ionok csúsznak DE beszárítás a normál emésztés után, hosszú enzimes inkubáció... → veszteségek, nem specifikus hasítások

9
Hogyan jelölhetünk? iTRAQ ™ (Applied Biosystems) Nagyon ravasz jelölés! amino-csoportra (N-terminus, Lys) 4-féle stabil izotópos szerkezet, azonos additív tömeggel, de különböző fragmensekkel (m/z 114, 115, 116, 117) azonosítás és kvanti az MS-MS adatokból!
 4-féle stabil izotópos szerkezet, azonos additív tömeggel, de különböző fragmensekkel (m/z 114, 115, 116, 117) azonosítás és kvanti az MS-MS adatokból!.")
10
iTRAQ jelölés 4 mintát kvantitatíve megkülönböztetni. Minden peptidet jelöl, a módosítottakat is Egyszerű a mintaelőkészítés Jelnövelő hatású (szuperponálódik a 4-féle jel) 20%-nál kisebb standard deviáció Jobb minőségű CID spektrumok Poszt-transzlációs változásokat is lehet így mérni! Ross et. al. MCP 2004 DRÁGA
 20%-nál kisebb standard deviáció Jobb minőségű CID spektrumok Poszt-transzlációs változásokat is lehet így mérni. Ross et. al. MCP 2004 DRÁGA.")
11
Mi ebből a tanulság?/ Take home message Ha biológus vagy → eredj programozást és statisztikát tanulni Ha matematikus vagy → irány a biológia Ha proteomikával akarsz foglalkozni → tömegspektrometria sem árt

12
Mi ebből a tanulság?/ Take home message Mennyiben lehet biológiai folyamatokat statisztikailag/matematikailag leírni? A komputer gyors, nem hibázik, „tanulékony”, de hülye... Mi emberek ellenben képesek vagyunk felismerni a váratlant, az újat – feltéve, hogy használjuk az eszünket....

13
Minta kérdések Mitől függ, mennyire jó egy adatbázis? Mi az oka, hogy nincs tökéletesen működő adatbázis-lekereső szoftver? Miért van szükség proteomikára? Miért kell „jelölni” kvantitatív proteomikában? Miért előnyös egy olyan jelölés, ami minden peptidet módosít? És valóban módosít-e minden peptidet?

14
Hasznos web-oldalak ProteinProspector – http://prospector.ucsf.eduhttp://prospector.ucsf.edu MS-BLAST - http://dove.embl- heidelberg.de/Blast2/msblast.htmlhttp://dove.embl- heidelberg.de/Blast2/msblast.html BLAST - http://www.ncbi.nlm.nih.gov/BLAST/http://www.ncbi.nlm.nih.gov/BLAST/ European Bioinformatics Institute - http://www.ebi.ac.uk/ http://www.ebi.ac.uk/ Szekvencia-összehasonlítás – http://www.ebi.ac.uk/clustalw/index.html MS kezdőknek - „What is mass spectrometry” - www.asms.org www.asms.org

Hasonló előadás














 Készítette: Bolla Zsuzsanna Környezetmérnök MSc.>")




