Előadást letölteni
Az előadás letöltése folymat van. Kérjük, várjon

1
Polycystas vesebetegségek klinikuma és genetikája
Prof. Dr. Túri Sándor SZTE Gyermekklinika

2
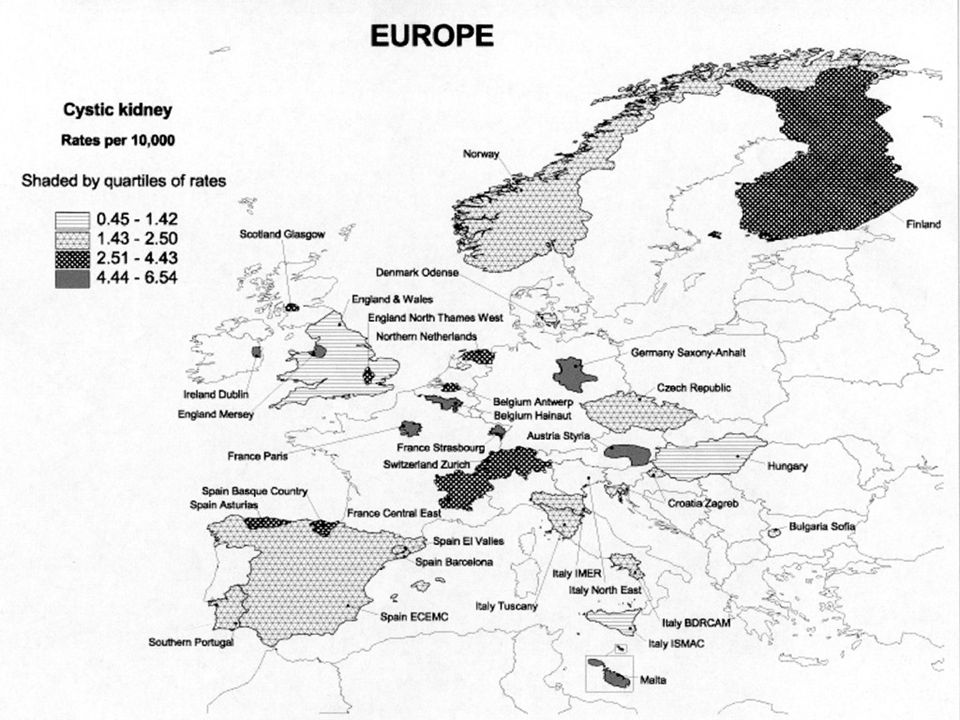
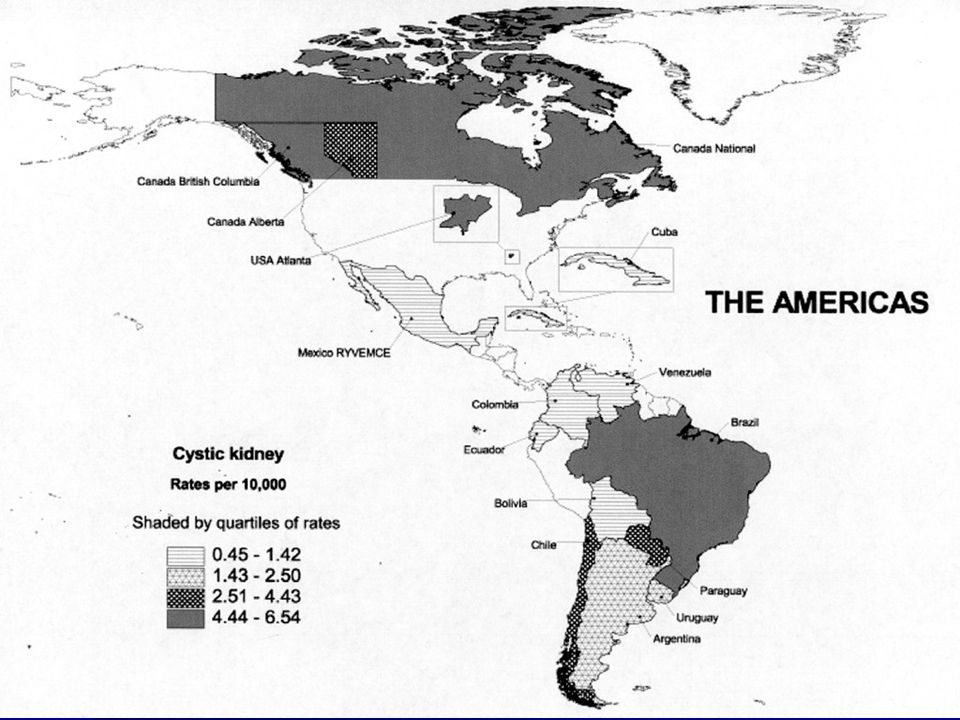
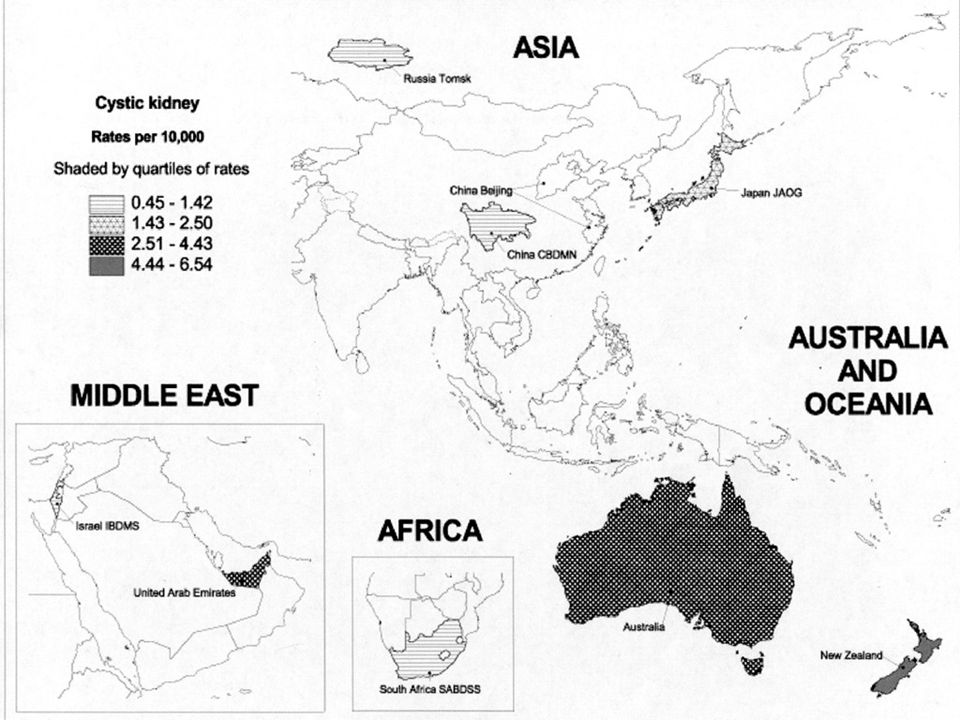
Az autoszóm domináns polycystas vesebetegség (ADPKD) a leggyakoribb monogénes nephropathia (1-2,5‰), általában felnőttkorban manifesztálódik, de kb. 2%-ban már gyermekkorban is jelentkezhet. elsősorban a vesék érintettek, gyakoriak a máj, pancreas cystak, cerebralis aneurismák. A dializáltak kb. 8-10%-át adják. Kapcsoltsági vizsgálatok genetikai heterogenitást jeleztek. Az autosom recesszív polycystas vesebetegség (ARPKD) egyike a legjelentősebb öröklődő nephropathiáknak gyermekkorban. Klinikai képe változatos, a súlyos korai gyermekkori formától az enyhébb, késői gyermekkori formáig terjedően. Incidenciája 1: Európában, penetranciája 100%. Génlocusa: 6p21-p12. Újabb neve: PKHD1: polycystic kidney and hepatic disease 1.
 egyike a legjelentősebb öröklődő nephropathiáknak gyermekkorban. Klinikai képe változatos, a súlyos korai gyermekkori formától az enyhébb, késői gyermekkori formáig terjedően. Incidenciája 1: Európában, penetranciája 100%. Génlocusa: 6p21-p12. Újabb neve: PKHD1: polycystic kidney and hepatic disease 1.")
3
PKD1 típus (16p13. 3, 46 exon) a betegek kb. 80-85%-ánál mutatható ki
PKD1 típus (16p13.3, 46 exon) a betegek kb %-ánál mutatható ki. Korábbi kezdet, agresszívebb klinikai lefolyás jellemzi, veseelégtelenség kb. 45%-nál ~50 év körül jelentkezik. 10% sporadikus, 90% familiáris. A PKD2 típus (4q21-23, 15 exon) kb %-nál igazolódott. Veseelégtelenség gyakran: 70 év. Fehérje termékeik (polycystin-1 és –2) egy receptor/ion-csatorna komplex részei. Néhány esetben eddig ismeretlen gén szerepel. Az autoszóm recesszív polycystas vesebetegség (ARPKD, 6p21-p12) nagyságú, 86 exonból áll, polyductin/fibrocystin.
 a betegek kb %-ánál mutatható ki. Korábbi kezdet, agresszívebb klinikai lefolyás jellemzi, veseelégtelenség kb. 45%-nál ~50 év körül jelentkezik. 10% sporadikus, 90% familiáris. A PKD2 típus (4q21-23, 15 exon) kb %-nál igazolódott. Veseelégtelenség gyakran: 70 év. Fehérje termékeik (polycystin-1 és –2) egy receptor/ion-csatorna komplex részei. Néhány esetben eddig ismeretlen gén szerepel. Az autoszóm recesszív polycystas vesebetegség (ARPKD, 6p21-p12) nagyságú, 86 exonból áll, polyductin/fibrocystin.")
4
Adult domináns polycystas vesebetegség (ADPKD)
Előfordulás 1:1000, egyik leggyakoribb örökletes betegség Autoszomális domináns öröklődésmenet A tünetek felnőttkorban manifesztálódnak Nagy polycystas vese >50% végstádiumú vesebetegség (ESRD) Több szervet érintő betegség (máj ciszták, cerebrális aneurizmák) Genetikailag heterogén (PKD1, PKD2)
 Több szervet érintő betegség (máj ciszták, cerebrális aneurizmák) Genetikailag heterogén (PKD1, PKD2)")
5
Klinikai diagnózis Ultrahang Computer tomographia MRI

6
ADPKD normál vese polycystas vese

8
Polycystas vese (ADPKD)
")
9
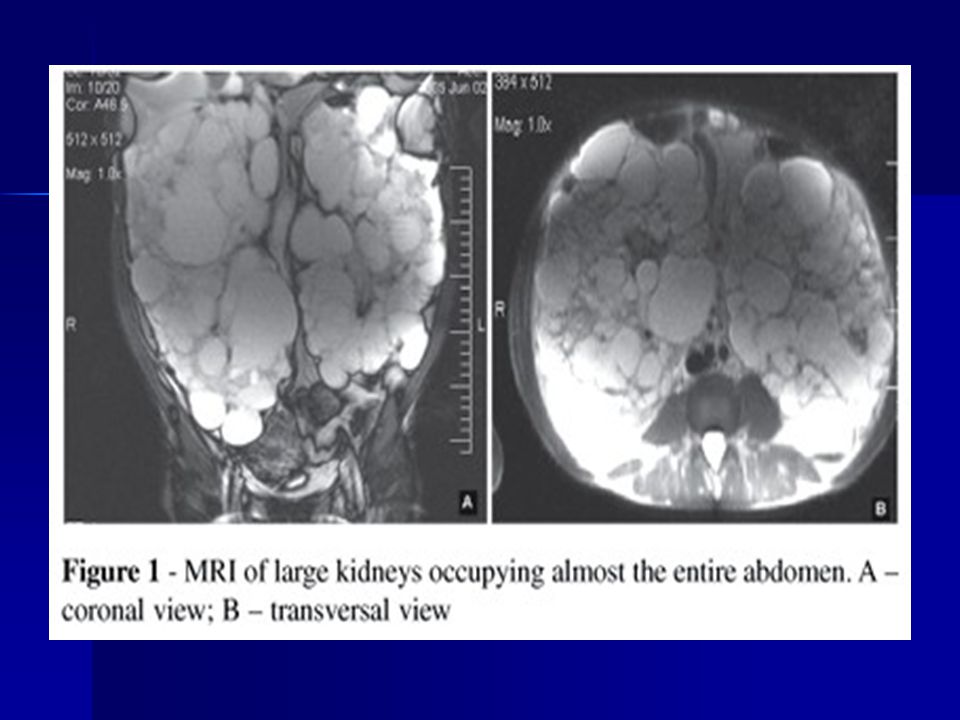
ADPKD kétoldali óriás polycystás vese

13
A polycystin-1 (PC1) és polycystin-2 (PC2) fehérjék lokalizációja
A PC1 és PC2 tansmembrán fehérjék Kálcium csatornaként funkcionálnak

14
A polycystin-1 (PC1) és polycystin-2 (PC2) fehérjék lokalizációja a vesében
PC PC2

15
Pathomechanizmus Károsodott intracelluláris Ca homeostasis,
fokozott cAMP, MAPK és mTOR szignál fokozott sejtproliferáció és apoptozis adja a terápiás kisérletek alapját. Vasopressin (V2) receptor antagonisták Somatostatin analogok a gerincesek Gs és a Gi receptorain hatva gátolják a cAMP, OPC31260 szintézisét (a gerincesek V2 receptora) Tolvaptan (a human V2 receptor erős antagonistája) effektív volt ezekben a modellekben és a V2 receptor expressziójának csökkentése mellett kevéssé toxikus. A fázis 3 Tolvaptan klinikai trial jelenleg is megy. A hoszú hatású somatostatin analog gátolja a patkányok vese és máj cystás betegségét PCK patkányokban. Egy pilot study szerint hatásos human ADPKD-ban is és további klinikai vizsgálatok vannak folyamatban.
 receptor antagonisták. Somatostatin analogok a gerincesek Gs és a Gi receptorain hatva gátolják a cAMP, OPC31260 szintézisét (a gerincesek V2. receptora) Tolvaptan (a human V2 receptor erős antagonistája) effektív. volt ezekben a modellekben és a V2 receptor expressziójának. csökkentése mellett kevéssé toxikus. A fázis 3 Tolvaptan klinikai trial jelenleg is megy. A hoszú hatású somatostatin analog gátolja a patkányok vese és máj. cystás betegségét PCK patkányokban. Egy pilot study szerint hatásos. human ADPKD-ban is és további klinikai vizsgálatok vannak. folyamatban.")
16
Mikor a GFR kezd csökkenni, a kezelésnek már nincs értelme
NIH Consortium: MR mérések a vese volumenére a cysták méretére, GFR mérések mutációs analízis A vese volumen jó prediktor a vesefunkció csökkenésére, és a progresszió megítélésére Human PKD Study : ACE gátló ACE gátló+ARB hatás vizsgálata ADPKD-ben a vérnyomásra, a kimenetelre, a kreatininre és a halálozásra

17
Betegek és Módszer: 32 ADPKD-s család összesen 187 tagja, 2 ARPKD-s család összesen 13 tagja
Short Tandem Repeat (STR) marker vizsgálatok perifériás fehérvérsejtekből izolált DNS mintákon. A PCR-t gél-elektroforézis (AlfExpress) követte. PKD1 markerek: D16S663, KG8, D16S291. PKD2 markerek: D4S1563, D4S2462. ARPKD markerek: D6S436, D6S427, D6S1344, D6S245, D6S1714, D6S295. Olyan STR markerekkel végezzük a kapcsoltsági analízist, melyek a betegséget okozó génben vagy szegélyező régiójában találhatók.
 marker vizsgálatok perifériás fehérvérsejtekből izolált DNS mintákon. A PCR-t gél-elektroforézis (AlfExpress) követte. PKD1 markerek: D16S663, KG8, D16S291. PKD2 markerek: D4S1563, D4S2462. ARPKD markerek: D6S436, D6S427, D6S1344, D6S245, D6S1714, D6S295. Olyan STR markerekkel végezzük a kapcsoltsági analízist, melyek a betegséget okozó génben vagy szegélyező régiójában találhatók.")
18
A PKD marker vizsgálatok gyakorlati alkalmazása: I
A PKD marker vizsgálatok gyakorlati alkalmazása: I. megerősíti a PKD1 klinikai formát KG8: II II III III III IV.1. Két 16-os kromoszóma marker is megerősítette a betegség ezen kromoszómához köthetőségét, a III.2. családtag 32 éves korától dializált.

19
A PKD marker (D4S2462) vizsgálat gyakorlati alkalmazása: II
A PKD marker (D4S2462) vizsgálat gyakorlati alkalmazása: II. megerősítette a PKD2 formát I II II.2. III.1. III.2. III.3. III.4. IV.1. IV.2. IV.3. IV.4. Peritonealis dial. 72 éves kortól!
 vizsgálat gyakorlati alkalmazása: II. megerősítette a PKD2 formát. I.2. II.1. II.2. III.1. III.2. III.3. III.4. IV.1. IV.2. IV.3. IV.4. Peritonealis dial. 72 éves kortól!")
20
A PKD marker vizsgálatok gyakorlati alkalmazása: III. A II. 3
A PKD marker vizsgálatok gyakorlati alkalmazása: III. A II.3. anya csak gyermeke (III.3.) genotípusát akarja tudni (etikai kérdés!) D16S291 I.1. II II III III II II III.3. A genetikai vizsgálat jelezte II.3.-nál is a valószínűleg génmutációs 16-os kromoszóma jelenlétét, ezt viszont ő tudni nem szeretné!
 genotípusát akarja tudni (etikai kérdés!) D16S291 I.1. II.1. II.2. III.1. III.2. II.3. II.4. III.3. A genetikai vizsgálat jelezte II.3.-nál is a valószínűleg génmutációs 16-os kromoszóma jelenlétét, ezt viszont ő tudni nem szeretné!")
21
A PKD markervizsgálat gyakorlati alkalmazása IV
A PKD markervizsgálat gyakorlati alkalmazása IV. a PKD1-es forma miatt fontos: a III.6. is gyanazt a 16-os kr.-át hordozza, mint testvére (de egyelőre „csak” hypertoniás!) D16S291: III.6. III II II.6. D16S663: III III II II.6.
 D16S291: III.6. III.7. II.5. II.6. D16S663: III.6. III.7. II.5. II.6.")
22
A PKD markervizsgálat gyakorlati alkalmazása
A PKD markervizsgálat gyakorlati alkalmazása . a PKD1-es forma miatt fontos: a gravid II.3. hordozza-e ugyanazt a 16-os kr.-át, mint testvére (kiderült, nem!) I I II II II III III.2.
 I.1. I.2. II.1. II.2. II.3. III.1. III.2.")
23
A PKD markervizsgálat gyakorlati alkalmazása a PKD1-es forma miatt: a II. és III. generáció, mivel mindegyik tagja a mutáns 16-os kromoszómát örökölte, in vitro fertilisatiot kér pozitív családtervezéséhez! I II II II II III III III.2.

24
Autoszomális recesszív polycystas vesebetegség (ARPKD)
Az ARPKD ritka genetikai rendellenesség. Gyakorisága populációnként változó, 1/ lakos közötti. Tünetei születéstől a serdülő korig, ritkán felnőtt korban jelentkeznek. Rendszerint csecsemőkori betegség, ekkor súlyos vese- és májtünetek az okai a morbiditásnak és mortalitásnak (ezért újabb angol neve: PKHD1: polycystic kidney and hepatic disease-1). (A vesék szimmetrikusan megnagyobbodottak, a vese gyüjtőcsatornáinak és a distalis tubulusoknak fusiform dilatatioja, valamint a máj portális triászainak dysgenesise, portalis epeútproliferatio congenitalis fibrosissal kísérve észlelhető.)
. (A vesék szimmetrikusan megnagyobbodottak, a vese gyüjtőcsatornáinak és a distalis tubulusoknak fusiform dilatatioja, valamint a máj portális triászainak dysgenesise, portalis epeútproliferatio congenitalis fibrosissal kísérve észlelhető.)")
25
ARPKD (PKHD-1) Terápia: oki egyelőre nincs (génterápia?)! A legtöbb csecsemő újszülöttkorban meghal. Azoknak, akik túlélik a csecsemőkort, esélyük van elfogadható életmódra a serdülőkorban gondos orvosi felügyelet mellett!
! A legtöbb csecsemő újszülöttkorban meghal. Azoknak, akik túlélik a csecsemőkort, esélyük van elfogadható életmódra a serdülőkorban gondos orvosi felügyelet mellett!")
26
ARPKD (PKHD-1) Az ARPKD foetalis jelei: Az újszülött/csecsemőkorban diagnosztizált esetek kb. 50%-ánál már a magzati UH is jelezte a kórképet (nagy vesék, a 13. héttől hyperechogenitás). A 20. héttől: az amniális folyadék kevés, ill. hiányozhat (oligohydramnion), akárcsak a magzati hólyag feltöltődése. Korrekt ARPKD magzati diagnosztika: direkt mutációs analízissel: jelenleg nem lehetséges, indirekt kapcsoltsági vizsgálattal: igen!
. A 20. héttől: az amniális folyadék kevés, ill. hiányozhat (oligohydramnion), akárcsak a magzati hólyag feltöltődése. Korrekt ARPKD magzati diagnosztika: direkt mutációs analízissel: jelenleg nem lehetséges, indirekt kapcsoltsági vizsgálattal: igen!")
27
ARPKD (PKHD-1) Újszülöttkorban: Potter sy (mélyen ülő szemek, ellaposodott orr, kórosan rövid állkapocs csontok, alacsonyan ülő fülek, esetleg: végtag rendellenességek, és a kevés amniális folyadék miatti egyéb elváltozások) %-uk születéskor, vagy röviddel azután meghal. Újszülöttkori halálokok: elsősorban a fejletlen tüdővel (pulmonális hypoplasia) kapcsolatosak, nem a veseelégtelenséggel (a tüdő növekedéséhez elegendő amniális folyadék szükséges!). Súlyos légzési distress, igen nagy vesék miatti korlátozott diaphragma mozgás. Egyéb komplikációk: pneumothorax, atelectasia, meconium aspiratio, bacterialis pneumonia, surfactant deficiencia.
 %-uk születéskor, vagy röviddel azután meghal. Újszülöttkori halálokok: elsősorban a fejletlen tüdővel (pulmonális hypoplasia) kapcsolatosak, nem a veseelégtelenséggel (a tüdő növekedéséhez elegendő amniális folyadék szükséges!). Súlyos légzési distress, igen nagy vesék miatti korlátozott diaphragma mozgás. Egyéb komplikációk: pneumothorax, atelectasia, meconium aspiratio, bacterialis pneumonia, surfactant deficiencia.")
28
ARPKD (PKHD-1) Megnövekedett intraabdominális nyomás miatti csökkent táplálék felvétel, a kóros gastro-intestinális motilitás miatti kisebb absorptio és a krónikus, progresszív veseelégtelenség. Vese növekedési csúcs: 1-2 éves korban, stabilizálódás: 4-5 évesen! Az újszülöttkort túlélő ARPKD-s gyermekek: A ventilláció sikeres elősegítése szignifikánsan megnöveli a túlélés esélyeit! Az 5 éves életben maradési arány 80-95% is lehet az első 4 élethetet túlélőknek. A vesék gondozása, a hypertonia kezelése gyakran a felnőttkorig segítheti elő a túlélést!

29
Kidney of a 2-year-old ARPKD child
Kidney of a 2-year-old ARPKD child. Macroscopically, the organ displays multiple small cysts on its surface and is severely enlarged, measuring ~15 cm in its longitudinal axis . B, Drawing representing the diffuse and radial distribution of dilated collecting ducts throughout the cortex and medulla. C, Renal histology, showing dilation of collecting ducts

30
ARPKD (PKHD-1) ARPKD-s veseműködési rendellenességek:
Nagy mennyiségű, nem koncentrált vizelet, polyuria, polydipsia. A dehydratio nagyobb a kockázata: Elhúzódó lázas állapotokban, hányás, hasmenés esetén Ugyanezt okozzák: forró nyári napok, sport aktivitás, ez megelőzhető megfelelő vizfogyasztással! Hypertonia: kb. 80%-uknál, néhány hónapos kortól, gyakran súlyos. Ok: nem pontosan ismert (csökkent vese vérátáramlás? RAS aktiválódás?) Az életkilátást rontja! Hyponatraemia (a szabad víz kiválasztás zavara miatt?)
 Az életkilátást rontja! Hyponatraemia (a szabad víz kiválasztás zavara miatt )")
31
ARPKD (PKHD-1) genetikája
Az ARPKD-s betegek >99%-ánál egyetlen gén mutációi felelősek a tünetekért. Recesszív: vagyis mindkét szülőtől kell egy-egy mutációt örökölni a betegség jelenkezéséhez. A gén (helye: 6p21- p12) egyike a legnagyobb emberi géneknek, mely minimum 86 kódoló exonból áll membránprotein, a polyductin/fibrocystin. A gén mutációk szinte családonként változhatnak! Az egy-egy génmutációt hordozó „carrier” szülők egészségesek!
 egyike a legnagyobb emberi géneknek, mely minimum 86 kódoló exonból áll membránprotein, a polyductin/fibrocystin. A gén mutációk szinte családonként változhatnak! Az egy-egy génmutációt hordozó „carrier szülők egészségesek!")
32
A polyductin/fibrocystin fehérje sematikus ábrája

33
ARPKD (PKHD-1) genetikája
Recesszív öröklődés sematikus ábrázolása: Egy génmutációt hordozó, egészséges apa anya A szülői 6-os kromoszómák tovább öröklődésének lehetőségei: A magzat/újszülött lehetséges genotípusai N=normál PKHD-1 gént hordozó 6-os kromoszóma jele n =mutáns PKHD-1 gént hordozó 6-os kromoszóma jele

34
ARPKD (PKHD-1) genetikája
Recesszív öröklődés esélyei: Ha mindkét szülő hordozza az ARPKD gén 1-1 mutációját (carrierek): 25% az esélye annak, hogy ARPKD-s gyermekük foganjon, 50% az esélye, hogy egy génmutációt hordozó, egészséges magzatuk legyen, 25% az esélye, hogy egy génmutációt sem hordozó, egészséges legyen az utód
: 25% az esélye annak, hogy ARPKD-s gyermekük foganjon, 50% az esélye, hogy egy génmutációt hordozó, egészséges magzatuk legyen, 25% az esélye, hogy egy génmutációt sem hordozó, egészséges legyen az utód.")
35
ARPKD (PKHD-1) genetikája
Ha ARPKD-s beteg társa carrier lenne 1 génmutációra, 50%-os valószínűséggel foganna egy génmutációs carrier és 50%-os valószínűséggel ARPKD-s beteg utód. Két ARPKD-s betegnek csak ARPKD-s gyermekük lehet! Ha ARPKD-e betegnek születne utóda olyan személytől, aki nem hordoz ARPKD-a génmutációt, mindegyik leszármazottuk egy génmutációt hordozó carrier lenne.

36
Magzati diagnosztika ARPKD-s beteg családjában
III.1 kislány újszülöttkorban exitált ARPKD miatt, első beteg a családban! Családvizsgálat kérésükre a gén STR markereivel, mely jelzi a génmutációs 6. kromoszóma öröklődését a családban mindkét ágon (carrierek). Kiderül, hogy a beteg kislány nővére nem hordoz ARPKD génmutációt. A szülők újabb gyermekvállalásukhoz kérik a magzati diagnosztikát (12. gestacios héten chorion boholy vétel, DNS extrakció.
. Kiderül, hogy a beteg kislány nővére nem hordoz ARPKD génmutációt. A szülők újabb gyermekvállalásukhoz kérik a magzati diagnosztikát (12. gestacios héten chorion boholy vétel, DNS extrakció.")
37
Magzati diagnosztika eredménye ARPKD-s beteg családjában
D6S436 (5’-végi) D6S1344 (intragenikus, 3’-vég felé) D6S1714 (intragenikus, 5’-vég felé) II II III III III II II III.1. III.2. III II II III.1. III.2. III.3. Az 5’-végi D6S436-os és a két intragenikus marker (D6S1344 és D6S1714) informatívnak bizonyult a családban, és lehetővé tette a magzati diagnosztika elvégzését. Az anya újabb terhessége kapcsán mind a három informatív marker azt jelzi, hogy a magzati mintából izolált DNS ugyanazt az apai és anyai 6-os kromoszóma öröklődését mutatja, mint amelyiket az ARPKD miatt elhunyt kislányuk esetében láthattunk. Ezért a magzat nagy valószínűséggel (>99%) szintén ARPKD-t örökölt.
 D6S1344 (intragenikus, 3’-vég felé) D6S1714 (intragenikus, 5’-vég felé) II.2. II.3. III.1. III.2. III.3. II.2. II.3. III.1. III.2. III.3. II.2. II.3. III.1. III.2. III.3. Az 5’-végi D6S436-os és a két intragenikus marker (D6S1344 és D6S1714) informatívnak bizonyult a családban, és lehetővé tette a magzati diagnosztika elvégzését. Az anya újabb terhessége kapcsán mind a három informatív marker azt jelzi, hogy a magzati mintából izolált DNS ugyanazt az apai és anyai 6-os kromoszóma öröklődését mutatja, mint amelyiket az ARPKD miatt elhunyt kislányuk esetében láthattunk. Ezért a magzat nagy valószínűséggel (>99%) szintén ARPKD-t örökölt.")
39
Polycystás vese MRI felvétele

40
Polycystás vese pathologiai képe

41
A polycystás vesebetegségben érintett gének és fehérje termékeik
PKHD1 PKD2 PKD1 hibás gén 6p21-23 4q21 16p13.3 locus receptor (?) ioncsatorna receptor funkció 4074 As 968 As 4302 As méret fibrocystin polycystin-2 polycystin-1 fehérje kb 5.6 kb 14.5 kb mRNS 86 15 46 exon 469 kb 110 kD 462 kD
 ioncsatorna. receptor. funkció As. 968 As As. méret. fibrocystin. polycystin-2. polycystin-1. fehérje kb. 5.6 kb kb. mRNS exon. 469 kb. 110 kD. 462 kD.")
Hasonló előadás




















>")